The Functional Role of the Hemoglobin-Water Interface
Abstract
The interface between hemoglobin (Hb) and its environment, in particular water, is of great physiological relevance. Here, results from in vitro, in vivo, and computational experiments (molecular dynamics simulations) are summarized and put into perspective. One of the main findings from the computations is that the stability of the deoxy, ligand-free T-state (T0) can be stabilized relative to the deoxy R-state (R0) only in sufficiently large simulation boxes for the hydrophobic effect to manifest itself. This effect directly influences protein stability and is operative also under physiological conditions. Furthermore, molecular simulations provide a dynamical interpretation of the Perutz model for Hb function. Results from experiments using higher protein concentrations and realistic cellular environments are also discussed. One of the next great challenges for computational studies, which as we show is likely to be taken up in the near future, is to provide molecular-level understanding of the dynamics of proteins in such crowded environments.
1 Introduction
The human red blood cell (RBC, erythrocyte) contains a complex aqueous
solution of hemoglobin, nonhemoglobin proteins, lipids, glucose,
electrolytes (mainly K+, Na+, Cl-, HCO, and phosphates)
and other components. Approximately 97 % of its volume is occupied
by water and hemoglobin and that of intracellular water alone of 72
%.1 Hence, the main interface between hemoglobin and
its intracellular environment is with water. Thus, it is essential to
be able to describe the interaction between Hb and water for
understanding the physiological function of Hb in RBCs.
In this contribution an overview of the current knowledge of the
Hb/water interface is provided with an emphasis on the structure and
dynamics of the protein and its environment. The solution properties
of hemoglobin were already reviewed by Antonini and Brunori in their
book published fifty years ago.2 At the time the
solution structure of the protein, its oligomerization state and the
behaviour in solutions with different ionic strengths were of
particular interest. A protein’s solvent environment and its chemical
composition can change its effects from acting as plasticizers
(e.g. water) to being stabilizers (e.g. trehalose or glycerol). At a
molecular level this is further complicated by the fact that protein
side chains can switch between conformational substates and that
certain such motions can be prevented. For example, for lysozyme it
was shown experimentally that the internal dynamics is activated when
the environment changes from pure glycerol (which is a stabilizer) by
increasing the level of hydration, because water is a
plasticizer.3 More generally it has been found
that protein dynamics is slaved to solvent
fluctuations.4 Increased hydration has also been
found to affect internal motions.5 Hence, it is
essential to better understand the interplay between solvent structure
and dynamics and the protein dynamics coupled to it and whether this
influences the biological function in a physiological context.
A first dynamical transition of the internal dynamics of proteins
between “low amplitude motion” and more “diffusive motion” occurs
between 180 K and 220 K6 although for the protein
thaumatin a transition at 110 K has been reported.7
This “glass transition” was investigated by using quasi-inelastic
neutron scattering experiments or Mössbauer
spectroscopy.8 Experiments on Hb in RBCs have reported
an “elastomeric transition” between a gel- and a fluid-like
phase.9 At a temperature of 310 K, human RBCs were
found to undergo a sudden change from blocking to passing through
micropipettes. This behavior was associated with a gel-to-fluid phase
transition9 which is also related to the known
increase in viscosity of highly concentrated Hb. Subsequent
experiments on RBCs from different organisms revealed that such a
transition involves Hb in all cases and that, more surprisingly, the
transition temperature is correlated with the body temperature of the
respective species.10
It was hypothesized that the drop in viscosity with subsequent changes
for cellular passage through micropipettes is caused by protein
aggregation.9 Concomitantly, Hb shows a pronounced
loss of its helical content at body temperature, not only for
human Hb, but also for several other species.11 It was
proposed12 that this is due to an increased amplitude
of the sidechain motions. This triggers unfolding of the helical
structure accompanied by an increase in surface hydrophobicity, which
eventually leads to protein aggregation. Experimentally, this was
investigated by temperature-dependent incoherent quasielastic neutron
scattering on whole red blood cells.10 Such
experiments are able to separate global and internal protein
motions. It was found that the amplitudes of protein side-chain
motions increased close to the body temperature of different species,
such as monotremes (305 K) and humans (311 K) with the difference due
to amino acid substitutions.
2 The Role of Water for the Stability of Hemoglobin
Hemoglobin (Hb) is one of the most widely studied proteins due to its
essential role in transporting oxygen from the lungs to the
tissues. Binding of molecular oxygen, O2, at the heme-iron is the
physiologically relevant step for oxygen transport. Oxygen
homeostasis13 at the molecular level can be described
as the dynamic equilibrium between Obound and ligand-free Hb at
one of the four heme-iron atoms and depends on the local O2
concentration. The self-regulation of oxygen homeostasis is reflected
in protein allostery,14 the capacity of proteins such as
Hb to modulate their affinity towards a physiological target (here
O2) by structural adaptations upon binding or removal of another
ligand (here O2) at a different binding site. In other words,
allostery is the molecular embodiment of homeostasis at the cellular
level. The two most important structural states of Hb are the deoxy
structure (T0), which is stable when no ligand (subscript “0”) is
bound to the heme-iron, and the oxy structure (R4), which is stable
when each of the four heme groups have a ligand (subscript “4”),
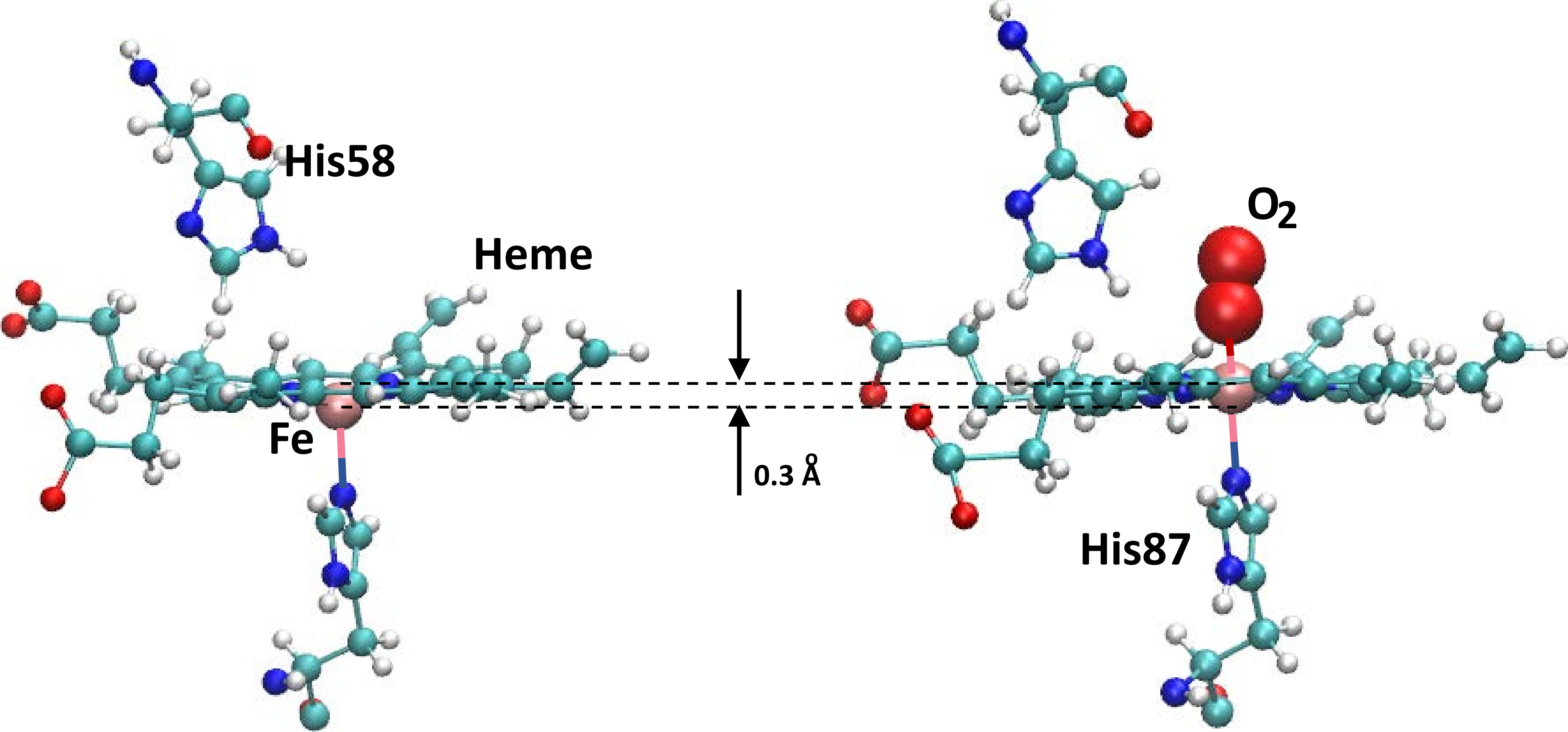
such as oxygen, bound to them, see Figure 1 for the
structure of the active site including the heme, the surrounding
histidine residues, and the bound O2 ligand (right panel). The
state with the quaternary structure of R4, but with no heme-bound
ligands is the R0 state. Despite strong experimental
evidence15 that T0 is significantly more stable
than R0, with an equilibrium constant of , molecular dynamics (MD) simulations appeared to indicate
that the R0 state is more stable than T0. Specifically,
simulations started with hemoglobin in its T0 state have been found
to undergo a spontaneous transition into the R0 state on sub-s
time scales.16, 17 Understanding the molecular
details of this discrepancy between the experimentally measured and
simulated relative stabilities of the R0 and T0 states is
essential for establishing the reliability of simulation-based studies
of Hb and other large biomolecules.
In a recent set of MD simulations it was found that the T0
R0 transition rate depends sensitively on the size of
the simulation box.18 The simulations were initialized
with Hb in the T0 state and immersed in a periodically replicated
cubic solvent box with side lengths of 75 Å, 90 Å, and 120
Å. In such boxes transitions towards the R-state structure were
observed after 130 ns, 480 ns, and 630 ns, respectively. By contrast,
in a water box with side-length 150 Å (top row Figure
2), Hb remained in its T0 state for the entirety of a
1.2s simulation. Extrapolation of this trend suggests that T0
is the thermodynamically stable state in this water box.
The results also suggested that such a large box is required for the
hydrophobic effect, which stabilizes the T0 tetramer, to be
manifested. Hydrophobicity is the tendency of a solute (here Hb) to
pack with itself and to exclude water molecules. In other words, the
solute and water segregate which leads to maximization of hydrogen
bonds between water molecules while minimizing the contact area
between the solute and water. The hydrophobic effect is one of the
main driving forces in the formation of biological interfaces,
including cell membranes and vesicles and thus is essential for
life.19
The importance of the hydrophobic effect as an organizing “force” in
biological systems has been recognized for quite some
time. Hydrophobicity has been discussed and established as an
important driver in protein folding or for the compartmentalization of
cells.19 However, the hydrophobic effect is also
prevalent in everyday life (in the function of detergents or
emulsions), in materials sciences, adhesion, and
confinement.20 One of the findings particularly
relevant to the present discussion is the dependence of hydration of a
hydrophobe on its size, see Figure 2 bottom
row. Considering an idealized spherical cavity of radius it was
shown that the density of water a distance from the surface of the
cavity depends sensitively on its size. For a small cavity (
Å, the size of methane, CH4) the water density adjacent to the
cavity is larger by a factor of two than the bulk density of water
because water attempts to maintain a H-bonding network as strong and
dense as possible.20 This changes with increasing
size of the cavity. For a cavity the size of hemoglobin (
Å) solvent density around the protein is depleted and approaches
bulk density asymptotically without evident structure in the radial
distribution function, , see bottom Figure 2. These
findings are consistent with earlier MD simulations of hydrophobic
hydration of melittin.21 In these simulations it was
found that depending on the local curvature of the protein (e.g. a
“flat” sheet region compared with a “pointed” turn),
hydrophobic residues of even the same chemical type show different
water hydration shells due to local constraints on hydrogen bonding
which lead to different orientational water structures. In other
words: flat surfaces impose different constraints on the water
H-bonding patterns than surfaces with large curvature. The predicted
overall behaviour of the radial distribution function from model
studies, as shown in the bottom panels of Figure 2, is
consistent with those found from the atomistic simulations for Hb, see
middle panel of Figure 2.

These findings demonstrate that the global and local structure of the
solute also influence the global and local hydration which, in turn,
affect the thermodynamic stability of the solute. Because the root
mean squared difference between the T0 and the R4 structures of
Hb is Å, their hydration is also expected to differ.
This supports the observed dependence of the thermodynamic stability
of T0 vs. R0, where R0 is expected to be R4-like, on the
size of the water box as found from the MD
simulations.18 While the statistical significance of
these findings has been a topic of recent discussion in the
literature,22, 23 the dynamic stability of the
T0 state exhibits a clear and systematic dependence on the size of
the solvent box. The differences in the degree of hydration between
the T- and the R-states is also consistent with earlier work based on
individual X-ray structures.24 It is well established
that the and interfaces in Hb
are large and closely packed. Although the allosteric transition has
been shown to be more complex,25 it can be
described as a rotation of the
dimer relative to the dimer. The TR
transition also involves breaking of several salt bridges, in accord
with the Perutz mechanism.26, 27
Concomitantly, the buried surface of the R-state is reduced by Å2 as compared with the T-state. Based on the relationship
between solvent accessible surface area and the associated hydrophobic
contribution to the free energy,28 burial of
additional protein surface will differentially stabilize the T-state
relative to the R-state. Hence, the decrease in the protein surface
exposed to the solvent suggests that hydrophobic effects should
stabilize the T-state. Further analysis is required to provide
conclusive evidence of the role of the hydrophobic effect and to
reveal the mechanistic origin of the dependence of the thermodynamic
stability of the T0 state, relative to the R0 state, on the
simulation box size. A related interesting and challenging aspect
concerns the question of whether the solvent water follows the
conformational TR transition or whether water drives this
transition. It is also possible that during different phases of this
transition the roles of solvent and solute switch or that it is more
meaningful to consider the solvent-solute system as a whole.

It is of interest to note that recent work on the A peptide,
which is the main component of amyloid plaques involved in Alzheimer’s
disease, found that different solvent box sizes yield similar radius
of gyration, secondary structure, intrapeptide, and peptide-water
hydrogen bonds.30 However, of considerable interest for
the present discussion, it was reported that the hydrophobic surface
area and exposure of the backbone conformations depend significantly
and sensitively on the solvent box size, irrespective of the force
field used in the simulations.30 Similarly, MD studies
of nanoparticle diffusion also reported differences in the hydration
depending on the size of the solvent box.31 For Hb as the
solute the water diffusion coefficients also depends on the size of
the simulation box, see Figure 3. It is found that with
increasing box size the diffusivity of water for the system including
hemoglobin approaches that of the solvent alone. Finally, following a
tangential approach to the problem by use of quasichemical theory it
was reported that both hydrophilic and hydrophobic contributions to
hydration depend on system size. They are predicted to decrease with
increasing system size. The net hydration free energy benefits
somewhat from the compensation of hydrophilic and hydrophobic
contributions which is akin to entropy/enthalpy
compensation.32, 33 Nevertheless, a large
system appears necessary to describe correctly the balance of these
contributions to the hydration of the
macromolecule.34
More molecularly resolved and quantitative work has been done to
understand the local hydration of Hb in its T0 and R0
states.35 For this, the local hydrophobicity (LH) was
evaluated36, 37 along MD trajectories of the
two conformational states in differently sized water boxes. The local
hydrophobicity can be viewed as a generalization of a radial
distribution function in that it provides information about both the
presence and orientation of water molecules at an
interface. Such an analysis for the T0 and R0 states of Hb found
that the breaking and formation of salt bridges at the and interface is accompanied by changes in
LH.
The above provides a molecular view of the Perutz mechanism which is
based on a two-state model involving an equilibrium between the T- and
the R-states.38, 26 In the Perutz
model the T0-state is “tense”, constrained by salt bridges
between the C-termini of the four subunits and it has a low oxygen
affinity with no ligands bound, whereas the R4-state is
“relaxed”, the salt bridges are broken and ligands are bound to the
heme-irons. The T0-state has the (5-coordinated) heme-iron out of
plane displaced towards the proximal histidine His87, see Figure
1, whereas in the R4 state the heme-iron is in the
plane and is six-coordinated with the ligand occupying the remaining
free valence as shown in Figure 1. Every heme-iron atom
is coordinated to a histidine residue at its distal site (“below the
heme plane”). The histidine residues are His87 in the chains
and His92 in the chains. Because these histidine residues are
part of the F-helix that is linked to the E-helix (containing the
proximal histidine residue) forming a loop, motion at the distal side
of each subunit can be efficiently transduced to other relevant parts
of the protein.39 The T0/R4 equilibrium it
thought to be governed primarily by the position of the iron atoms
relative to the porphyrin and the salt bridge stability and dynamics
is considered to be linked to the distal
histidines.26, 27 This interplay between
ligand binding, local and global conformational changes, the change in
interface exposed to the solvent and subsequent rearrangement of
solvent leads to a logical chain of events spanning various length
scales (from atomic to mesoscopic) that govern Hb function.
For Hb in cubic water boxes with 90 Å and 120 Å edge length it
was found that simulations initialized in the T0 state decay to
known but different intermediate structures upon destabilization of
the interface following a decrease in LH; i.e. as a
consequence of reduced water density or change of water orientation at
the protein/water interface.35 This is in line with
earlier simulations18 that reported a reduced number of
water-water hydrogen bonds in smaller simulation boxes which shifts
the equilibrium between water-protein and water-water contacts and
changes the activity of water. Interestingly, for decreasing box
sizes, simulations for the A peptide reported a pronounced
increase in the hydrophobic surface area30 and studies
of protein G found an increase in the hydrophobic contribution due to
a decrease in solvent density fluctuations as the system size
decreases.34 This is consistent with studies for Hb
that found larger hydrophobic exposure in smaller simulation
boxes.35
3 Characterization of the Protein/Water Interface from Measurements on RBCs
In contrast to studies of individual macromolecules, such as Hb, in
aqueous solution containing adequate concentrations of anions and
cations (see previous sections), proteins in a realistic cellular
environment experience extreme crowding.40 Hence,
although the above studies are of interest to better understand the
physical behaviour of complex macromolecules at a molecular level,
they are not necessarily directly relevant to the behaviour of
proteins under physiological conditions.
A notable experiment41 determined the
diffusivity of water in Hb/water mixtures at protein concentrations of
mg/mL, which is representative of the crowded cellular
environment with protein concentrations of up to 400 mg/mL. Using
quasielastic incoherent neutron scattering (QENS) to probe the water
dynamics of RBCs in D2O and H2O buffer, the cytoplasmic dynamics
of H2O was separated from membrane and macromolecular dynamics. The
difference in the two signals is dominated by the water dynamics
because hydrogen nuclei have an incoherent scattering cross section
that is times larger than that of macromolecules or
deuterium. The translational diffusion of cellular water was found to
be nearly identical to that of H2O buffer which is consistent with
the results from MD simulations if sufficiently large solvent boxes
are used, see Figure 3. However, this finding is
surprising for cellular environments for three reasons. First, the
average separation of macromolecules in a crowded cellular environment
is of the order of 10 Å which corresponds to only layers
of water molecules. Second, NMR experiments42 and MD
simulations43 have found that the reorientation
dynamics of water on the protein surface is slowed down by a factor of
2 to 3 compared with water in the bulk. Third, time resolved
fluorescence spectroscopy reported that a significant fraction of the
water molecules is slowed down by an order of
magnitude.44 Although in fact, the notion that the
dynamics of water adjacent to a protein surface differs from that in
the bulk dates back at least 60 years,45 there is as
yet no explanation for the high diffusivity of cellular water.
Much effort has gone into characterizing the behaviour of Hb in
RBCs.46 Of particular relevance is the
understanding of the sensitivity of the volume of RBCs to changes in
the osmolality of the surrounding medium. Such effects were referred
to as “anomalous osmotic behaviour” of RBCs.47
Interestingly, the molecular explanation for the apparent anomaly is a
cooperative effect by which the total charge of Hb decreases with
increased Hb concentration.48
4 Relevance of In Vitro Studies to Physiology
As noted above, in a cellular environment the spatial separation
between proteins is of the order of 10 Å. This differs
considerably from most MD studies that investigate one or a few
proteins in solution. Similarly, NMR experiments are carried out under
dilute conditions that avoid clustering of the proteins. Hence, the
question arises in what sense such in vitro studies are relevant
to the situation encountered in vivo.
A first obvious difference, as already mentioned, is the “crowding”
encountered in real cells. Typically, physico-chemical experiments aim
at reducing the complexity in order to obtain specific
information about the system of interest - here the energetics and
dynamics of Hb. However, crowding and the presence of multiple
interaction partners generally makes the interpretation of experiments
on RBCs difficult. For example, when using infrared spectroscopy to
probe site-specific dynamics in a protein much effort is spent in
finding molecules that absorb in a frequency range that is largely
devoid of responses from the protein.49 This range
extends from cm-1 to cm-1. Hence,
spectroscopic probes such as -CN,50
-SCN,51 or
N352, 53, 54 which absorb between
2000 cm-1 and 2300 cm-1 are ideal reporters since all
signals in this frequency range can be unambiguously assigned to the
reporter groups.
One significant study directly probed the water dynamics in a cellular
environment.55 Contradicting the view that a
substantial fraction of cell water is strongly perturbed, it was found
that % of cell water in E. coli and in the extreme
halophile Haloarcula marismortui had bulk-like dynamics,
consistent with the results in Figure 3. The remaining
% of cell water interacts directly with biomolecular
surfaces and is motionally retarded by a factor on average,
corresponding to a rotational correlation time of 27 ps. This dynamic
perturbation is three times larger than for small monomeric proteins
in solution, a difference that was attributed to secluded surface
hydration sites in supramolecular assemblies.
More recently, all atom MD simulations of the Mycoplasma
genitalium (Mg), the simplest bacterium, with a genome of 470 genes
versus E. coli, which has about 4600 genes, have been
performed.56 They included all molecular components
(i.e. proteins, RNA, metabolites, ion, and water) explicitly in
atomic detail with a total of million atoms. The system
was simulated for 20 s. Even with this short simulation time some
interesting results were obtained; e.g., partial denaturation due to
protein-protein interactions occurred and macromolecular diffusion was
slowed down.
Another example of the differences between in vivo and in
vitro studies was found for the production of recombinant
adeno-associated viruses.57 Although this example is
not related to hemoglobin per se, it nicely illustrates that the
behaviour of a complex system (such as a multimeric protein) depends
on the structure and composition of its environment (i.e., in a cell
or under idealized laboratory conditions). For the production of such
viruses, all conditions were maintained identical except for the host
cell species in which they were grown. Interestingly, the post
translational modifications of the viruses expressed in the two
different cell types differed and the sites at which methylation
occurred also depended on the cell line that was used. It was
concluded that virus receptor binding, trafficking, or expression
kinetics can depend on the method used to grow the virus.
5 Summary
The present work highlights the importance of a molecular-level
understanding of the interface between hemoglobin and its environment,
notably water, under in vivo and in vitro conditions. In
cellular environments, such as RBCs, crowding is the main difference
when compared with more idealized realizations of a system in
vitro in most computational and physico-chemical
experiments. Although following such “divide-and-conquer” approaches
has provided remarkable insights into the dynamics and thermodynamics
of complex systems, their relevance to those experienced under
physiological conditions needs to be examined in detail. The finding
that 85 % of the water in cells behave like bulk water and only 15 %
has slowed its diffusion by one order of magnitude suggests that
laboratory experiments and molecular simulations with slight
modifications (“crowders”) should be able to emulate many aspects of
real-cell environments without unduly increasing the complexity of the
system investigated. This is even more likely to be a meaningful
approach as simulations for Hb carried out in sufficiently large
solvent boxes confirm these findings. Bridging the gap between
idealized single-molecule scenarios typically considered in current
laboratory and simulation-based experiments and the crowded cellular
environments is a challenge that will be taken up in the near future
to obtain a molecular-level understanding of complex biological
systems, including living cells.
Acknowledgment
The authors thank Adam Willard for insightful comments and thoughtful
correspondence. Support by the Swiss National Science Foundation
through grants 200021-117810, the NCCR MUST (to MM), and the
University of Basel is acknowledged. The support of MK by the CHARMM
Development Project is gratefully acknowledged.
References
- Levin et al. 1976 Levin, R.; Cravalho, E.; Huggins, C. Effect of hydration on the water content of human erythrocytes. Biophys. J. 1976, 16, 1411–1426
- Antonini and Brunori 1971 Antonini, E.; Brunori, M. Hemoglobin and Myoglobin in Their Reactions with Ligands; North-Holland Publ. Co., Amsterdam, 1971
- Paciaroni et al. 2002 Paciaroni, A.; Cinelli, S.; Onori, G. Effect of the environment on the protein dynamical transition: a neutron scattering study. Biophys. J. 2002, 83, 1157–1164
- Fenimore et al. 2002 Fenimore, P. W.; Frauenfelder, H.; McMahon, B. H.; Parak, F. G. Slaving: solvent fluctuations dominate protein dynamics and functions. Proc. Natl. Acad. Sci. 2002, 99, 16047–16051
- Smith et al. 1989 Smith, J.; Kuczera, K.; Tidor, B.; Doster, W.; Cusack, S.; Karplus, M. Internal dynamics of globular proteins: comparison of neutron scattering measurements and theoretical models. Physica B: Condensed Matter 1989, 156, 437–443
- Doster et al. 1989 Doster, W.; Cusack, S.; Petry, W. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature 1989, 337, 754–756
- Kim et al. 2011 Kim, C. U.; Tate, M. W.; Gruner, S. M. Protein dynamical transition at 110 K. Proc. Natl. Acad. Sci. 2011, 108, 20897–20901
- Achterhold et al. 2002 Achterhold, K.; Keppler, C.; Ostermann, A.; Van Bürck, U.; Sturhahn, W.; Alp, E.; Parak, F. Vibrational dynamics of myoglobin determined by the phonon-assisted Mössbauer effect. Phys. Rep. 2002, 65, 051916
- Artmann et al. 1998 Artmann, G.; Kelemen, C.; Porst, D.; Buldt, G.; Chien, S. Temperature transitions of protein properties in human red blood cells. Biophys. J. 1998, 75, 3179–3183
- Stadler et al. 2008 Stadler, A. M.; Digel, I.; Artmann, G.; Embs, J. P.; Zaccai, G.; Buldt, G. Hemoglobin dynamics in red blood cells: correlation to body temperature. Biophys. J. 2008, 95, 5449–5461
- Zerlin et al. 2007 Zerlin, K. F. T.; Kasischke, N.; Digel, I.; Maggakis-Kelemen, C.; Artmann, A. T.; Porst, D.; Kayser, P.; Linder, P.; Artmann, G. M. Structural transition temperature of hemoglobins correlates with species’ body temperature. Eur. Biophys. J. 2007, 37, 1–10
- Digel et al. 2006 Digel, I.; Maggakis-Kelemen, C.; Zerlin, K.; Linder, P.; Kasischke, N.; Kayser, P.; Porst, D.; Artmann, A. T.; Artmann, G. Body temperature-related structural transitions of monotremal and human hemoglobin. Biophys. J. 2006, 91, 3014–3021
- Semenza 2010 Semenza, G. L. Oxygen homeostasis. Wiley Interdisciplinary Reviews: Systems Biology and Medicine 2010, 2, 336–361
- Cui and Karplus 2008 Cui, Q.; Karplus, M. Allostery and cooperativity revisited. Prot. Sci. 2008, 17, 1295–1307
- Edelstein 1971 Edelstein, S. Extensions of the allosteric model for haemoglobin. Nature 1971, 230, 224–227
- Hub et al. 2010 Hub, J. S.; Kubitzki, M. B.; de Groot, B. L. Spontaneous Quaternary and Tertiary T-R Transitions of Human Hemoglobin in Molecular Dynamics Simulation. PLoS. Comput. Biol. 2010, 6, e1000774
- Yusuff et al. 2012 Yusuff, O. K.; Babalola, J. O.; Bussi, G.; Raugei, S. Role of the Subunit Interactions in the Conformational Transitions in Adult Human Hemoglobin: An Explicit Solvent Molecular Dynamics Study. J. Phys. Chem. B 2012, 116, 11004–11009
- El Hage et al. 2018 El Hage, K.; Hedin, F.; Gupta, P. K.; Meuwly, M.; Karplus, M. Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size. eLife 2018, 7, e35560
- Tanford 1978 Tanford, C. The hydrophobic effect and the organization of living matter. Science 1978, 200, 1012–1018
- Chandler 2005 Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647
- Cheng and Rossky 1998 Cheng, Y.; Rossky, P. Surface topography dependence of biomolecular hydrophobic hydration. Nature 1998, 392, 696–699
- Gapsys and de Groot 2019 Gapsys, V.; de Groot, B. L. Comment on ‘Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size’. eLife 2019, 8, e44718
- El Hage et al. 2019 El Hage, K.; Hedin, F.; Gupta, P. K.; Meuwly, M.; Karplus, M. Response to comment on ‘Valid molecular dynamics simulations of human hemoglobin require a surprisingly large box size’. eLife 2019, 8, e45318
- Lesk et al. 1985 Lesk, A.; Janin, J.; Wodak, S.; Chothia, C. Hemoglobin - The surface buried between the and dimers in the deoxy and oxy structures. J. Mol. Biol. 1985, 183, 267–270
- Fischer et al. 2011 Fischer, S.; Olsen, K. W.; Nam, K.; Karplus, M. Unsuspected pathway of the allosteric transition in hemoglobin. Proc. Natl. Acad. Sci. 2011, 108, 5608–5613
- Perutz 1970 Perutz, M. F. Stereochemistry of Cooperative Effects in Haemoglobin. Nature 1970, 228, 726–734
- Perutz et al. 1998 Perutz, M. F.; Wilkinson, A.; Paoli, M.; Dodson, G. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Ann. Rev. Biophys. Biomol. Struc. 1998, 27, 1–34
- Chothia 1974 Chothia, C. Hydrophobic bonding and accessible surface area in proteins. Nature 1974, 248, 338–339
- Yeh and Hummer 2004 Yeh, I.; Hummer, G. System-size dependence of diffusion coefficients and viscosities from molecular dynamics simulations with periodic boundary conditions. J. Phys. Chem. B 2004, 108, 15873–15879
- Mehra and Kepp 2019 Mehra, R.; Kepp, K. P. Cell size effects in the molecular dynamics of the intrinsically disordered A peptide. J. Chem. Phys. 2019, 151
- Cui and Cui 2021 Cui, A. Y.; Cui, Q. Modulation of Nanoparticle Diffusion by Surface Ligand Length and Charge: Analysis with Molecular Dynamics Simulations. J. Phys. Chem. B 2021,
- Sharp 2001 Sharp, K. Entropy-enthalpy compensation: Fact or artifact? Prot. Sci. 2001, 10, 661–667
- Chodera and Mobley 2013 Chodera, J. D.; Mobley, D. L. Entropy-enthalpy compensation: role and ramifications in biomolecular ligand recognition and design. Ann. Rev. Biophys.. 2013, 42, 121–142
- Asthagiri and Tomar 2020 Asthagiri, D.; Tomar, D. S. System size dependence of Hydration-Shell occupancy and its implications for assessing the hydrophobic and hydrophilic contributions to hydration. J. Phys. Chem. B 2020, 124, 798–806
- Pezzella et al. 2020 Pezzella, M.; El Hage, K.; Niesen, M. J.; Shin, S.; Willard, A. P.; Meuwly, M.; Karplus, M. Water dynamics around proteins: T- and R-States of Hemoglobin and Melittin. J. Phys. Chem. B 2020, 124, 6540–6554
- Willard and Chandler 2010 Willard, A. P.; Chandler, D. Instantaneous liquid interfaces. J. Phys. Chem. B 2010, 114, 1954–1958
- Shin and Willard 2018 Shin, S.; Willard, A. P. Characterizing hydration properties based on the orientational structure of interfacial water molecules. J. Chem. Theor. Comput. 2018, 14, 461–465
- Antonini and Brunori 1971 Antonini, E.; Brunori, M. Hemoglobin and Myoglobin in Their Reactions with Ligands; North-Holland Publ. Co., Amsterdam, 1971; pp Chapter 14, Section 14.3.4
- Kachalova et al. 1999 Kachalova, G. S.; Popov, A. N.; Bartunik, H. D. A steric mechanism for inhibition of CO binding to heme proteins. Science 1999, 284, 473–476
- Zhou et al. 2008 Zhou, H.-X.; Rivas, G.; Minton, A. P. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Ann. Rev. Biophys.. 2008, 37, 375–397
- Stadler et al. 2008 Stadler, A. M.; Embs, J. P.; Digel, I.; Artmann, G. M.; Unruh, T.; Buldt, G.; Zaccai, G. Cytoplasmic water and hydration layer dynamics in human red blood cells. J. Am. Chem. Soc. 2008, 130, 16852–16853
- Mattea et al. 2008 Mattea, C.; Qvist, J.; Halle, B. Dynamics at the protein-water interface from 17O spin relaxation in deeply supercooled solutions. Biophys. J. 2008, 95, 2951–2963
- Sterpone et al. 2012 Sterpone, F.; Stirnemann, G.; Laage, D. Magnitude and molecular origin of water slowdown next to a protein. J. Am. Chem. Soc. 2012, 134, 4116–4119
- Pal et al. 2002 Pal, S. K.; Peon, J.; Zewail, A. H. Biological water at the protein surface: dynamical solvation probed directly with femtosecond resolution. Proc. Natl. Acad. Sci. 2002, 99, 1763–1768
- Bernal 1965 Bernal, J. The structure of water and its biological implications. Symposia of the Society for Experimental Biology. 1965; pp 17–32
- Longeville and Stingaciu 2017 Longeville, S.; Stingaciu, L.-R. Hemoglobin diffusion and the dynamics of oxygen capture by red blood cells. Sci. Rep. 2017, 7, 1–10
- Savitz et al. 1964 Savitz, D.; Sidel, V. W.; Solomon, A. Osmotic properties of human red cells. J. Gen. Physiol. 1964, 48, 79–94
- Gary-Bobo and Solomon 1968 Gary-Bobo, C.; Solomon, A. Properties of hemoglobin solutions in red cells. J. Gen. Physiol. 1968, 52, 825–853
- Koziol et al. 2015 Koziol, K. L.; Johnson, P. J. M.; Stucki-Buchli, B.; Waldauer, S. A.; Hamm, P. Fast Infrared Spectroscopy of Protein Dynamics: Advancing Sensitivity and Selectivity. Curr. Op. Struct. Biol. 2015, 34, 1–6
- Zimmermann et al. 2011 Zimmermann, J.; Thielges, M. C.; Seo, Y. J.; Dawson, P. E.; Romesberg, F. E. Cyano Groups as Probes of Protein Microenvironments and Dynamics. Angew. Chem. Intern. Ed. 2011, 50, 8333–8337
- van Wilderen et al. 2014 van Wilderen, L. J. G. W.; Kern-Michler, D.; Mueller-Werkmeister, H. M.; Bredenbeck, J. Vibrational dynamics and solvatochromism of the label SCN in various solvents and hemoglobin by time dependent IR and 2D-IR spectroscopy. PCCP 2014, 16, 19643–19653
- Bloem et al. 2012 Bloem, R.; Koziol, K.; Waldauer, S. A.; Buchli, B.; Walser, R.; Samatanga, B.; Jelesarov, I.; Hamm, P. Ligand Binding Studied by 2D IR Spectroscopy Using the Azidohomoalanine Label. J. Phys. Chem. B 2012, 116, 13705–13712
- Salehi et al. 2019 Salehi, S. M.; Koner, D.; Meuwly, M. Vibrational Spectroscopy of N3–in the Gas and Condensed Phase. J. Phys. Chem. B 2019, 123, 3282–3290
- Salehi and Meuwly 2021 Salehi, S. M.; Meuwly, M. Site-selective dynamics of azidolysozyme. J. Chem. Phys. 2021, 154, 165101
- Persson and Halle 2008 Persson, E.; Halle, B. Cell water dynamics on multiple time scales. Proc. Natl. Acad. Sci. 2008, 105, 6266–6271
- Yu et al. 2016 Yu, I.; Mori, T.; Ando, T.; Harada, R.; Jung, J.; Sugita, Y.; Feig, M. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. eLife 2016, 5, e19274
- Rumachik et al. 2020 Rumachik, N. G.; Malaker, S. A.; Poweleit, N.; Maynard, L. H.; Adams, C. M.; Leib, R. D.; Cirolia, G.; Thomas, D.; Stamnes, S.; Holt, K. Methods matter: standard production platforms for recombinant AAV produce chemically and functionally distinct vectors. Mol. Therap. - Meth. Clin. Devel. 2020, 18, 98–118