On the algorithm to perform Monte Carlo simulations in cells with constant volume and variable shape
Abstract
In simulations of crystals, unlike liquids or gases, it may happen that the properties of the studied system depend not only on the volume of the simulation cell but also on its shape. For such cases it is desirable to change the shape of the box on the fly in the course of the simulation as it may not be known ahead of time which geometry fits the studied system best. In this work we derive an algorithm for this task based on the condition that the distribution of specific geometrical parameter observed in simulations at a constant volume matches that observed in the constant-pressure ensemble. The proposed algorithm is tested for the system of hard-core ellipses which makes lattices of different types depending on the asphericity parameter of the particle. It is shown that the performance of the algorithm critically depends on the range of the sampled geometrical parameter. If the range is narrow, the impact of the sampling method is minimal. If the range is large, inadequate sampling can lead to significant distortions of the relevant distribution functions and, as a consequence, errors in the estimates of free energy.
Key words: hard-ellipse fluid, Monte Carlo simulation, constant volume, varying shape, umbrella sampling
Abstract
 ñèìóëÿöiÿõ êðèñòàëiâ ïåâíi âëàñòèâîñòi äîñëiäæóâàíî¿ ñèñòåìè ìîæóòü çàëåæàòè íå òiëüêè âiä îá’єìó êîìiðêè ìîäåëþâàííÿ àëå é âiä ¿¿ ôîðìè.  òàêèõ âèïàäêàõ áàæàíî çìiíþâàòè ôîðìó êîìiðêè â ïðîöåñi ñèìóëÿöié, îñêiëüêè íàïåðåä âîíà ìîæå áóòè íåâiäîìîþ. Ó öié ðîáîòi îïèñàíî àëãîðèòì ÿêèé äîçâîëÿє öå ðîáèòè ç òî¿ óìîâè, ùîá âiäòâîðèòè êëþчîâi ïàðàìåòðè ôîðìè êîìiðêè, ÿêi ñïîñòåðiãàþòüñÿ â àíñàìáëi ïðè ïîñòiéíîìó òèñêó. Àëãîðèòì ïðîòåñòîâàíî â ñèìóëÿöiÿõ ñèñòåìè òâåðäèõ åëiïñiâ, ÿêà ìîæå óòâîðþâàòè ãðàòêè ðiçíîãî òèïó. Ïîêàçàíî, ùî âèêîðèñòàííÿ çàïðîïîíîâàíîãî àëãîðèòìó ïðèçâîäèòü äî äîáðîãî óçãîäæåííÿ âiäíîñíî¿ âiëüíî¿ åíåðãi¿ ðiçíèõ òèïiâ ãðàòîê ç ðåçóëüòàòàìè îòðèìàíèìè íåçàëåæíèìè ìåòîäàìè.
Ключовi слова: òâåðäèé åëiïñ, Ìîíòå Êàðëî, àíñàìáëü ïðè ïîñòiéíîìó îá’єìi
1 Introduction
Today computer simulations play a key role in fundamental research across multiple disciplines, including physics, chemistry and materials science [1]. A large share of computational studies employ simulation boxes with fixed volume. This choice is mainly motivated by convenience as constant-volume/constant-temperature ensemble is easier to program than the equivalent ensemble with constant pressure. But this is also due to the involvement of the constant-volume ensemble in other, specialized simulation techniques such as free energy calculations [2], Gibbs ensemble [3] or replica-exchange method [4, 5, 6]. Regardless of the particular context, it is always understood that the effect of volume vanishes in the thermodynamic limit where the results are thought to be independent of the employed ensemble. This claim is certainly true for liquids or gases, whose properties are independent of the geometry of the box.
In the case of crystals, however, the situation could be quite different [7, 8]. In crystalline materials there could be properties that depend explicitly on the volume as well as on the shape of the box. Take for instance the example of a rectangular lattice with lattice constants and , as shown in figure 1. The dimensions of the box that accommodates columns and rows are along axis and along axis as shown in figure 1 (a). The corresponding aspect ratio is . Now, let us assume that the lattice constants are not known ahead of time but are meant to be determined in the course of the simulations. If we initially choose a box with the wrong aspect ratio, for instance, see figure 1 (b) for appropriate illustration, the lattice constant determined in simulations will also be incorrect. Since the geometry of the cell drives the structure of the lattice, wrong geometry translates into wrong structure. Importantly, lattice distortions will not go away easily even when the size of the simulation box is increased.

One way to deal with this issue is to estimate the free energy of the studied system as a function of some geometrical parameter of the cell, for instance , and then choose the geometry with the lowest . This can be done by a variety of tools, including the Einstein crystal method for the free energy computation [9, 2]. Although formally correct, this approach is cumbersome and carries a large computational cost. An alternative is to allow the shape of the box to change in the course of the simulation. The proper geometry then corresponds to the free energy minimum, so it will be seen as the most frequently visited structure. An additional benefit will be for systems that can populate multiple geometries at the same time as their relative free energy in this case can be determined from a single Monte Carlo (MC) trajectory.
The question then is how does one allow the shape of the box to change? How is that accomplished in practice? Would, for instance, generating randomly, from time to time, a new aspect ratio in the course of the simulation constitute a good method? If not, what is the good method? These questions had been addressed before as multiple studies report using simulation cells with constant volume but variable shape (MCVS) [10, 11, 7, 8]. Unfortunately, the details of the performed simulations are scarce and to establish the specifics of the used algorithms appears difficult. Yet, we find evidence that the method one employs to sample the trial geometries may have measurable consequences for physical properties extracted from simulations. Thus, which geometry parameters are best to choose in MCVS simulations, and how to choose them remains unclear. These are the questions that we answer in the present article. We show that in order for the constant-volume ensemble with variable shape to be consistent with the constant-pressure ensemble, the aspect ratio should be sampled from the distribution. Any other sampling law will lead to erroneous results. We illustrate this point for the system of impenetrable ellipses in two-dimensional space, for which we evaluate the performance of the method that relies on uniformly sampled , or on the so-called -sampling and show that it produces wrong free energy for the relevant states of the system.
2 Theory
2.1 Sampling law for
Let us assume that, in addition to volume, the partition function in the canonical, or NVT for short, ensemble explicitly depends on some geometrical parameter , for instance the aspect ratio of the box sides. Here, is the inverse temperature, is the volume, is the potential energy and is the abbreviation for the point in the configuration space. The integration is carried out over volume with the set parameter . The full partition function then should be constructed as a weighted sum (or integral) over all possible realizations of the additional degree of freedom [12]. In the most general case, one finds:
| (2.1) |
where is the weighting function of the extended ensemble defined by both volume and the shape of the box and is the probability distribution function of , characteristic of the NVT ensemble with variable shape. In principle, one is free to choose arbitrarily, provided that it satisfies certain conditions typically imposed on distribution functions, such as positive definiteness or integrability. Here, we will select on the condition that the extended ensemble with fixed volume satisfies distribution of specific for the constant pressure, or NPT, ensemble. In this way, modelling in the two ensembles, constant-volume and constant-pressure, will be consistent, hence minimizing finite size effects.
The distribution function in the constant-pressure ensemble reads:
| (2.2) |
where is the pressure. Let us focus now on 2D space and obtain formulas for this simpler case first. The relevant volume is , where and are the lengths of the simulation box and is the sine of the angle between them. In NPT simulations, volume is sampled randomly from a uniform distribution. This can be achieved either by uniformly sampling one of the variables involved in volume, , or , or by non-uniformly sampling some combination of these variables which leads to a uniformly distributed volume [2]. Let us assume that the first scenario takes place, as it is more general and can be applied to both liquids and crystals, and each concerned variable is sampled uniformly, one at a time and in random order. The joint distribution function measured in such simulations for variables , and will be given by the following expression:
| (2.3) |
which can also be obtained by integrating distribution function (2.2) over all configurations . The dependence on in the partition function arises because the integration over is carried out for the box with specific dimensions and , which in addition to the volume also define other geometrical parameters including .

As an illustration, consider a schematic distribution shown in figure 2, specific for rectangular boxes with . Among all possible configurations, those that correspond to volume satisfy the constraint , creating a one-dimensional sub-ensemble characterized by a single degree of freedom. If is chosen as the independent degree of freedom, configurations with given and will appear in simulations with probability . Any other geometric parameter of the system will also be characterized by a unique distribution function. This includes the aspect ratio , for which the distribution function represents the relative frequency of seeing conformations with different . This function is directly accessible in simulations.
Our goal is to evaluate and then try to reproduce it in simulations at a constant volume. Using as the starting point, can be evaluated as , where the brackets indicate a statistical average over sub-ensemble with fixed . The constant-volume constraint can be imposed through another delta function, leading to the following expression in terms of the unbiased distribution function:
| (2.4) |
This integral can be evaluated in two steps. First, a change of variables helps to eliminate the integration over while keeping the other variable constant. The result is:
| (2.5) |
The second integration can be carried out by making a change of variables , which after dropping the terms that are independent of yields:
| (2.6) |
Let us now require that , i.e., the distribution functions in the NPT and NVT ensembles are equal. It is easy to see then from equation (2.1) that , indicating that . In other words, we arrive at the conclusion that new aspect ratios in constant-volume simulations should be drawn from the distribution in order to reproduce the result of the NPT simulations. Furthermore, it is seen from equation (2.6) that free energy associated with the degree of freedom can be computed as . Therefore, the aspect ratio is not suitable for the computation of free energy differences directly from the distribution function. In other words, , where and are some values defining two macroscopic states. It is easy to show, however, that is the distribution function for a new variable . Thus, in terms of this variable , making the proper order parameter associated with the geometry parameter . In the Appendix we show that sampling law also applies in the three-dimensional space.
2.2 Simulation algorithm
Given that the sampling law is known, how does one conduct a constant-volume simulation with the variable shape? Let us first point out the following auxiliary results. The lengths of the box can be expressed in terms of and volume when the cell angle is considered constant: and . Given that the volume is fixed, one can find the appropriate distributions for the lengths using the standard identity: , where is some function of and is the distribution function of this variable. It can be shown that they are given by the same expression as for : , where In other words, the sampling distributions of the size in and directions obey the same law. This result is of fundamental importance. The invariance with respect to the swap of and coordinates is central to simulations of condensed matter (with the exception of cases where external fields are imposed that break the symmetry). Any algorithm that violates this condition should be considered flawed. For instance, uniform sampling of assumes that , and so one can find that while . Both distributions are incorrect and will lead to a bias in the sampled ensemble. Another point that should be made regarding the law concerns its interpretation. Since small ’s correspond to narrow boxes while large ones — to wide boxes, the decline of the distribution function with may seem to indicate that the balance between the two types of boxes is broken. This impression, however, is misleading and the number of generated narrow and wide boxes is actually the same. The number of the former can be estimated as , where is some small range in which is allowed to vary. If, instead, one varies , the same number of boxes should be . Now, let us swap and coordinates in the last expression and obtain . This operation is expected not to affect the number of boxes, so one should find that . After some rearrangement, it follows that the condition for the balance between narrow and wide boxes is . It is easy to see that the derived formula satisfies this condition, proving that the symmetry between different shapes is preserved.
Up to this point we implicitly assumed that can be changed by changing either or . It is easy to see, however, that when the volume of the box is kept fixed . Thus, it is possible to change the aspect ratio also by changing the cell angle . It is clear that the distribution law should not depend on how is changed. We conclude, therefore, that the function should also apply for the angle moves. It is also clear from the expression of the volume that the term can be treated on the same footing as that of . Thus, one can bypass the derivation and immediately conclude that the sampling probability . The pertinent order parameter for the cell angle is . Accordingly, free energy associated with can be computed as .
It is convenient to combine the two types of moves, one in which and change simultaneously and one in which changes together with either or , in one algorithm that consists of two steps:
-
1.
A new value for is generated from distribution.
-
2.
A decision is made randomly with equal probability about which step is to take next: a) new , or b) new . The coordinates of the particles are appropriately scaled by and in the and directions in trial moves. Changes of the cell angle do not affect the coordinates.
Generating distributions can be achieved in a variety of ways. Probably, the easiest is the Metropolis importance sampling [9]. It consists in using a uniformly distributed random variable to generate a trial that is then accepted with probability .
3 Model and methods
3.1 Mathematical model
We test the designed algorithm for the system of hard-core ellipses. Ellipses have long and short half-axes and , respectively. Aspect ratio is defined as . It is a key parameter determining the general behavior of the system. The geometrical details are explained in figure 3 (a).

There are particles in the system. For the sake of generality we use a skew reference frame. Each particle is characterized by three degrees of freedom: two coordinates and one rotation angle , see figure 3 (b) for illustration. In addition to the skew coordinates, one can also assign Cartesian coordinates to each particle; the two are related as follows:
| (3.1) |
The volume elements of the two coordinates systems are related through the Jacobian of transformation . The particles are placed in a box with sides and and an angle between them under periodic boundary conditions, as shown in figure 3 (b)–(c). The skew simulation boxes are designed to accommodate lattices of all possible types. The minimum image convention is applied to compute the interaction energy. As illustrated in figure 3 (c), certain particle interacts with the closest periodic image of particle . The distance between the two particles is computed as , where and . The usual rule for computing the shortest distance is used: if , then it is replaced by . Similarly, if , then it is replaced by . The same transformations are applied to the coordinate . The Cartesian coordinates of the vector connecting two particles are and . Together with the rotation angles and , they are used to determine whether two ellipses overlap [13]. The density of the system is reported in reduced units where is the density of the maximally compact lattice configuration.
3.2 Relative free energy
To measure free energy difference between prospective lattice structures, we use the properly defined order parameters. As discussed in the section 2, one such parameter is , where . The ratio of the side lengths is measured directly in simulations and then binned to compute the associated distribution.
Another order parameter that will be utilized is . The rational why the cell angle can be employed as an order parameter is as follows. The system of ellipses can exist either as a fluid or a solid lattice phase, depending on the density [14]. At [15, 16], ellipses in the solid phase are not aligned with one another, making the so-called plastic lattice [13]. At larger aspect ratios, the particles are aligned similarly to nematic fluids. An example of the simulation cell in which ellipses are aligned is shown in figure 4 (a). The depicted close-packed conformation can be generated from the minimal motif that contains 4 ellipses. It is shown in panel (b) and also highlighted in the right-hand lower corner of the cell. This motif can be obtained from a close-packed configuration of disks by a sequence of unique steps. Let us assume that the diameter of the disks is and the angle that the left side of the initial box makes with the vertical axis is , see figure 4 (e). Let us rotate the box counterclockwise by an angle , as shown in figure 4 (d). After that let us stretch the box by the amount in the vertical direction. As a result, disks are transformed into ellipses. The length of the long axis of the ellipses becomes , as shown in figure 4 (c). Simultaneously, stretching changes the angle that the lower side of the box makes with the horizontal axis, as figure 4 (c) illustrates. While initially it was , now the angle becomes
| (3.2) |
The angle between the left-hand side and the vertical axis also changes. Immediately following the rotation, it is but after stretching it becomes
| (3.3) |
As can be seen from figure 4, physically distinct configurations are generated when changes between 0 and . All other values lead to redundant configurations. To characterize the state of each ellipse in a specific lattice state, one can use, for instance, the angle that the long axis makes with the horizontal axis, . As can be seen from figure 4 (b) . The geometry of the box, on the other hand, can be uniquely specified by its angle (given that the aspect ratio is fixed). Both angles, and , are functions of , so there is only one independent variable that fully defines the close-packed structure. All other parameters can be expressed as functions of the chosen variable via relations (3.2) and (3.3). For instance, the rotation angle can be cast as a function of :
| (3.4) |
where is the aspect ratio. For the purpose of illustration, figure 4 (e) shows this function for , .

It can be shown that varies in the range , while the corresponding limits of the rotation angle are . As changes, the structure of the lattice changes with it, creating the manifold of an infinite number of lattices differing from one another by the rotation angle . This is in contrast to the system of disks, which makes a single hexagonal lattice at high densities. As will become clear below, the lattice types corresponding to the extreme points have a special meaning. At , see figure 4 (a), the lattice can be viewed as being assembled from vertical lines of ellipses that make contact with one another through the pole at the short side of the particle. The ellipses are aligned along the line connecting their centers. Consequently, the corresponding lattice type is termed longitudinal (L). The lattice observed at can be assembled from horizontal lines of ellipses which contact each other through the pole at their long side. The orientation of particles is perpendicular to the line connecting their centers, so the corresponding lattice type is called transverse (T). In simulations, the longitudinal lattice can be transformed into the transverse lattice and vice versa by varying the angle . If is treated as a variable, the transition will happen spontaneously. The system in this case will sample various types of lattices in the course of a single simulation, thereby enabling the computation of their statistical weight. A major difficulty associated with this approach is convergence. To collect sufficient statistics for the distribution function , the system should visit different lattice types a large number of times, which may be difficult if the free energy difference between the concerned lattices is large. To improve the convergence, in this study we employ the method of umbrella sampling [17]. The range is broken equidistantly into a number of bins, or windows, each assigned a distinct value of . Harmonic potential is applied to bias sampling in simulations to the vicinity of each window. The unbiased distribution is reconstructed by collecting information on sampled in all windows. The order parameter relevant for the cell angle is .
3.3 Method of Einstein crystal
We also compute free energy differences by the method of Einstein crystal (EC) [18, 19]. The key idea of this method is to obtain free energy of the lattice states of interest relative to a common reference state. The free energy of the reference state then drops when the free energy difference is taken. The reference state is modelled by the harmonic Hamiltonian . Free energy with respect to this model is computed by thermodynamic integration with the help of a coupling constant . How this scheme works in skew coordinates is described in detail in the Appendix. The main formula we use to compute the free energy difference between lattice states characterized by different ’s is:
| (3.5) |
Here, is the average of the potential energy difference computed in simulations driven by the “hybrid” Hamiltonian . The actual potential energy of the hard-ellipse system is and the simulations are performed at a fixed volume and cell angle in the reference frame associated with the center of mass. As is varied between 0 and 1, the Hamiltonian is transformed from to . As the harmonic potential gradually becomes weaker, the integral in equation (3.5) reports on the spatial extent by which the system is allowed to deviate from the initial configuration, thus providing a measure of the configurational freedom.
The reference Hamiltonian
contains a set of coordinates with respect to which the free energy is evaluated. As a reference we chose a lattice configuration and ; , . Here, and are the dimensions of the simulation cell in and direction, are initial angles and and are adjustable spring constants. It is easy to show that , where is the ratio of the and dimensions. Thus, is uniquely defined by , , and . Parameter was extracted from constant-volume simulations as the average over all sampled cell side ratios. The integral in equation (3.5) was evaluated by numerical quadrature. In order to reduce the variation seen in , a non-uniform transformation was applied before integration. A total of 33 grid points, , were generated non-uniformly between and . The points were chosen so as to maintain a constant level of the numerical integration error, which, after optimization, we judge to be negligible. The statistical error was estimated by performing 5 independent measurements for each state point from which standard deviation was extracted.
3.4 Numerical details
The performed MCVS simulations consist of two types of moves. The first type is the regular MC steps in which particles are randomly displaced and rotated. The maximum magnitude of these steps are adjusted to achieve greater than 30% acceptance. The second type of moves are changes of geometry. They are attempted randomly with 10% overall probability. Box lengths and box angle are updated as described in the previous section. Parameters of these moves are adjusted to achieve greater than 50% acceptance rate. Changes of box lengths are accompanied with the appropriate scaling of particle coordinates. The algorithm of Vieillard-Baron [13] is used to determine if two ellipses overlap.
To perform the umbrella sampling simulations, the range [,] was divided equidistantly into 50 windows,. Simulations were performed in each window with an additional harmonic potential applied. The strength of the potential was set such that the adequate overlap of the histograms at the neighboring windows was achieved. The unbiased distribution was obtained at the end of the simulations by the multiple-histogram reweighting method [20, 21].
4 Results
We test the proposed algorithm in simulations of the system of hard-core ellipses. For the sake of comparison, we consider the scheme in which is sampled uniformly, i.e., the -sampling algorithm, in addition to the proper 1 sampling law. It is found that proper sampling is essential when the parameter controlling the geometry of the cell varies in a wide range. In other cases, different sampling methods lead to indistinguishable results.
4.1 Solid plastic phase
The first test was performed for a system with . We used rectangular boxes and set the density at which is high enough to trigger the transition into the crystalline state yet low enough to preclude aligning of the ellipses. Under these conditions, the system makes the so-called plastic lattice in which particles occupy lattice sites but are capable of rotating by all 180 degrees. A time trace of observed in simulations of this system is shown in figure 5 (a). The aspect ratio varies in the range around an average of ~1.15. At all times, the system occupies a lattice configuration with the distance between neighboring sites while the ellipses are capable of adopting any angle . A cartoon representation of this structure and its pair distribution function computed for the centers of the ellipses are shown in figure 5 (f) and figure 5 (e), respectively. The lack of alignment between particles is immediately apparent.

We find that the alignment (or rotational degrees of freedom more generally) plays a critical role in determining the structure of the lattice. When it is introduced manually by constraint where is the common angle of all particles which is allowed to change, the lattice splits into two sub-lattices with a distinct structure. One sub-lattice displays contacts between neighboring ellipses going through the pole at the long side and can be recognized as the transverse lattice. A cartoon representation of this lattice is shown in figure 5 (h). The second sub-lattice is the longitudinal lattice, exhibiting closest contacts between the neighboring ellipses going through their short side, as illustrated in figure 5 (g). All other lattice configurations correspond to free energy maxima and are not directly observable. It is not clear if this is a genuine property of the system or an artifact resulting from the use of rectangular box geometry, suppressing all structures other than the mentioned two. For the demonstration purposes, we considered a small system consisting of particles making lattice, so as to enable spontaneous T-L transitions. The time trace of obtained in simulations of this system is shown in figure 5 (b). It is seen that changes in a discontinuous manner among four different values, indicating that the geometry of the box is specific to the lattice type that it contains and suggesting that the aspect ratio can be used as a structural parameter to distinguish between T and L states. Pair distribution functions obtained for each lattice type, shown in figures 5 (c), (d), demonstrate that the two lattices have a noticeably different structure. The most obvious differences concern the first coordination shell. Compared to the model with full particle rotations, in the T and L states is split into two sub-maxima. The position of the first sub-maximum in T conformations is at while in L conformations it is at , demonstrating that the particles in the transverse arrangement are capable of approaching each other at shorter distances. This is understandable, given how ellipses are stacked in the two structures, see figure 5 (g) and (h). The second sub-maximum appears at in both states. The second and third coordinate shells also show significant differences between T and L conformations.
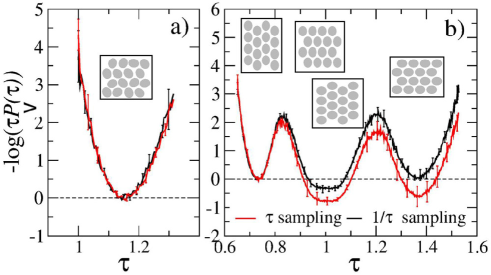
So how well does the proposed algorithm reproduce the statistics of the different conformational states? Figure 6 shows the free energy profile obtained for the system where all particles are free to rotate — panel a), and for the rotationally-constrained system — panel b). As can be expected from figure 5, a single minimum is seen for the unconstrained system and four minima are present for the constrained one. The specific aspect ratios of the minima are , , and . The two extreme values, and , correspond to the longitudinal state. The two minima in the middle are characteristic of the transverse state; they appear to merge in the graph because of being closely spaced. Conformations with and , and those with and , are related by a 90-degree rotation of the reference frame (note that no such transformation ever takes place in the simulations). Corresponding to the same state, they should exhibit the same free energy. It is a crucial test for the algorithm to reproduce this behavior. It is seen from figure 6 (b) that and within the error bars, so the proposed algorithm successfully passes the test. By contrast, the -sampling algorithm predicts that but and the difference is statistically significant. The greater population of state can be explained by the greater statistical weight assigned to conformations with larger by an algorithm in which the sampling probability is flat compared to the case when it declines with . Furthermore, even though the -sampling method generates the same free energy for and , the value it predicts is twice as large as that of the proper algorithm. Again, the difference is statistically meaningful, which leads us to the conclusion that this method is not capable of reproducing accurately the inter-state statistics for the constrained model under the considered conditions.
By contrast, figure 6 (a) shows that both sampling algorithms lead to the same results, within error bars, for the unconstrained system. It follows, therefore, that the performance of geometry sampling algorithms strongly depends on the system under study, more specifically on the range in which the geometry parameter is varied. If the range is narrow, as in figure 6 (a), the use of the proper algorithm does not make much difference. The average and the shape of the distribution function are generally well reproduced. The algorithm matters, however, when changes in a wide range, as shown in figure 6 (b). Improper sampling leads to significant distortions in that may ultimately affect the free energy estimates.
4.2 Free energy difference between distinct lattice types
When is increased beyond ~1.5, the ellipses in the crystalline state become aligned [15, 16]. What is the statistical weight of lattice types with different ellipse orientations? To find that out, we considered the system with . The cell angle for this system varies in the range [0.41, 1.43], which is much wider than the range [0.96, 1.12] appropriate for . This improves the chances of observing a quantitative difference between the results of using different sampling methods. We considered the cells with 6 rows and 6 columns of ellipses because smaller systems failed to produce stable lattices. Since spontaneous transitions among different lattice types were not observed for the considered system, we had to employ umbrella sampling method to compute the distribution function of the structural order parameter . The details are provided in the section 3. The density of the system was set at which is much higher than the density of the fluid-solid transition.

Free energy profile obtained in simulations using the proposed sampling algorithm is shown in figure 7 by black line. It has two minima with the lowest free energy corresponding to the transverse and longitudinal states. This agrees with our results for the constrained model of , suggesting that the presence of two identified stable states is not an artifact of the used cell geometry. The free energy of the longitudinal state is higher than that of the transverse state. The corresponding figure obtained by the -sampling method is . It is seen that this method assigns additional statistical weight to the longitudinal state with smaller . This is in line with our observation from the previous section where we saw states with larger experiencing more frequent sampling. Indeed, since , more sampling for large means more sampling for small and, by extension, small .
Free energy difference between the L and T states was also evaluated by the EC method. The geometry of the cell was determined in simulations using sampling. For the longitudinal structure we used and . For the transverse state, the corresponding numbers were , and . The initial ellipse orientations were generated by for both structures. Upon combining the results of 5 independent measurements, the free energy difference was evaluated as . This number is in excellent numerical agreement with the prediction made in the sampling simulations, , providing an essential validation for this method. By contrast, the -sampling algorithm generates an incorrect free energy, , and the error is statistically significant. Based on these observations, we conclude that the use of the proper sampling algorithm is essential for the studied system.
The free energy of the transverse state appears to be lower than that of the longitudinal state. This could be a genuine physical effect stemming from conformational preferences of different lattice configurations. Alternatively, the difference could be a by-product of the small size of the simulation cell. Simulations designed to extract free energy difference as a function of could help to resolve this ambiguity. Linear segments in at large would indicate genuine free energy difference between the two lattice states. Any other dependence would signal finite-size artifacts. Which of these two scenarios takes place needs to be answered in careful finite-size scaling studies.
5 Conclusions
We introduced an algorithm to perform MC simulations of crystalline systems in boxes with fixed volume but variable shape. Tests performed for the system of hard-core ellipses showed that the performance of the algorithm depends on the range in which the geometrical parameter characterizing the shape varies. If the range is narrow, which is probably the case for the majority of crystalline simulations, the use of the algorithm does not make much difference in comparison with other ad hoc sampling schemes. For wide ranges of the parameters, however, the use of the algorithm may be crucial. In the example of structural transition between transverse and longitudinal lattices, we find that the error in the free energy difference due to the inadequate sampling method reaches 40%. How large it may become, and how wide the corresponding variation range should be, will depend on the system of interest. However, as a general rule, the error due to the incorrect sampling should not be simply neglected.
At the same time, we note that the magnitude of the error declines when the range of the sampled parameter becomes narrower. This will happen, among other reasons, when the number of particles increases. Thus, proper finite-size analysis, in addition to yielding important information about the scaling properties of the studied system, will also help to combat the errors associated with the inadequate methods of sampling simulation box shapes.
Acknowledgements
The author expresses his utmost gratitude to the men and women of the Armed Forces of Ukraine, the National Guard and other law-enforcement agencies who made this study possible by their selfless service and, for many, ultimate sacrifice. The author would also like to thank Gerhard Kahl, Susanne Wagner, Roman Melnyk and Andriy Stelmakh for stimulating discussions during the time when this study was conceived and completed. This study was supported by the National Academy of Sciences of Ukraine, project KPKVK 6541230.
Appendix
A.1 Algorithm for the three-dimensional space
It is possible to conduct constant-volume simulations with the changing shape in the three-dimensional space. Let us assume that the geometry of the box is defined by six variables, three lengths and , and three angles and , as shown in figure A.1. Let us further assume for now that the box is rectangular, . The analogue of the constant-pressure distribution (2.3) in 3D will be a function of the lengths of the box:
| (A.1) |

Aspect ratios can be computed for all three sides of the box lying in , and planes. We will try to change only one at a time. The corresponding distribution function, for instance for , can be computed as follows:
| (A.2) |
By making the substitution one can eliminate the integral over obtaining:
After the second substitution and elimination of , one arrives at:
This result does not depend on the integration order. Therefore, we find that the sampling law in three-dimensional space is the same as in the two-dimensional space.
Since there is no difference with respect to the dimensionality, it is convenient to use the algorithms developed for 2D and apply them to 3D boxes. In particular, the idea of sampling one side of the box and then updating alternatively either another side or the angle between the sides can be reused. The volume of the box with variable angles is
In MC sampling, one should take alternatively pairs of a side and a second side or a side and an angle until all such combinations are exhausted. If the length is updated to then companion steps should be updating or . The second possible combination is (, or . The third and final combination is (, or . Specific formulas for how to update the lengths and angles can be obtained from the condition that the volume of the cell remains constant. If for instance is sampled and a trial value is , then the trial value for the length in the -direction should be and the trial angle in the plane
Similar formulas can be obtained for and combinations by permutations.
A.1.1 Einstein crystal method in skew coordinates
There are certain caveats associated with the use of EC method in conjunction with the skew coordinates, which we explain below. To evaluate the Hamiltonian, we need momenta in addition to the coordinates. These can be obtained using a general formula , where is the coordinate, is the time derivative of the coordinate and is the kinetic energy. For Cartesian coordinates, the kinetic energy is known: , where is the mass of the particle. It yields the familiar momenta and . Assuming that skew and Cartesian coordinates are related by expression (3.1) it follows that
| (A.3) |
thus, one can find the skew momenta: and . The reverse transformation and allows one to compute the Jacobian: leading to the volume element . The kinetic energy
can be found by expressing time derivatives of the coordinates in terms of the momenta and then substituting the results into equation (A.3).
Now, let us compute the partition function for one particle:
where we used the property that coordinates and momenta can be partitioned and U is the potential energy of the system. The two integrals in this expression correspond to ideal and excess partition functions giving rise to ideal and excess free energy, correspondingly. Let us evaluate the ideal part in the skew reference frame:
which turns out to be exactly the same as the equivalent expression in the Cartesian coordinates. Extending these calculations to particles will produce , which for sufficiently large will give the familiar expression of the free energy of ideal gas: , where . Note that this expression does not depend on , so the ideal part of the free energy can be neglected when working in the skew reference frame if relative free energy is of interest. Moving on to the excess partition function, one finds for one particle:
Generalization to particles
| (A.4) |
gives rise to the excess free energy , which is fully defined by the interaction energy .
Let us focus on the system of hard disks for now, and introduce a scaling constant that will transform the hard-sphere potential into the harmonic potential :
| (A.5) |
Here, are the positions of the ideal lattice with respect to which the free energy is evaluated. At , the system is described by the harmonic potential. It constitutes a set of independent harmonic oscillators with frequency controlled by the spring constant . At , we obtain the system of our interest — hard spheres. The “hybrid” potential defines some fictitious system which physically makes sense only at the extreme points of . The free energy of that fictitious system can be used to compute the free energy difference between the end points: . Since is known analytically, this formula allows us to compute as a sum , making the evaluation of a key task. This task can be accomplished by taking the derivative of with respect to :
| (A.6) |
where symbol denotes average in ensemble generated by . After the integration of the derivative, one obtains:
This is the key formula which relates the free energy of the system of interest to the integral obtained in simulations of the hybrid system:
| (A.7) |
Provided that is taken sufficiently large, the partition function in harmonic approximation can be evaluated analytically as:
The corresponding free energy then is:
where is the Boltzmann constant. In this expression, we singled out the second term that depends on . The first term will vanish in calculations of free energy differences using the same but different . Thus, the harmonic free energy relevant for our calculations is just . The final formula for the free energy becomes:
| (A.8) |
For ellipses, the methodology remains the same except that now we need to add new degrees of freedom — particle angles. The ideal part of the free energy will get new terms arising from new variables (comprising the inertia tensor of the ellipses) but these are independent of , so they can be neglected. In the excess part, we will get additional integration over the angles with normalization constant . The harmonic potential will now also apply to the angles:
where is an additional spring constant and are the set of initial particle angles. The new partition function changes into:
but the free energy part that depends on remains the same: .
In the course of simulations, we discovered that the integrand in formula (A.8) converges very inefficiently for , which corresponds to the system driven by the hard-core potential while its potential energy is evaluated with the help of the harmonic potential. The potential energy in this case experiences strong fluctuations that decay very slowly over time. After some research, the slow convergence was tracked down to the movements of the simulation cell as a whole. Since the random displacements of the particles during MC steps are uncorrelated, the center of mass of the system experiences displacements from its initial position as well. These displacements average out over time because of the law of large numbers but it may take a long simulation time in order to see that. For non-zero ’s, the collective movements of all particles are not an issue because the harmonic potential suppresses large-scale deviations from the initial coordinates. At the same time, the displacements of all particles as a whole do not create a physically distinct state and thus should not affect the relative free energy. It, therefore, makes sense to transition to the reference frame associated with the center of mass of the system. The description then includes the coordinates of the center of mass , and coordinates of the particles which characterize their mutual arrangement (conformations) (the coordinates of the remaining particle are expressed in terms of these new variables). Under periodic boundary conditions, the center of mass samples from the volume , where is the total volume of the system. This is the quantity that will appear in front of the integral (A.4) when the integration is carried out over and . Importantly, this volume per particle is the same for all , so the associated term of free energy will drop when the difference is taken. Integration over the remaining coordinates yields:
with the relevant -dependent term of free energy . This leads to the final expression (3.5) for the free energy given in the section 3. The function is evaluated in simulations carried out in the reference frame of the center of mass. In practice, this was achieved by aligning the coordinates of the center of mass at each MC step. We found that this can be accomplished by keeping track of two sets of coordinates: one containing real coordinates and the other storing coordinates that are periodically imaged. This prevented discontinuous jumps of the center of mass position when particles were put back into the simulation cell by the boundary conditions. We found that formula (3.5) produces the same free energy difference as formula (A.8) but at a much lower computational cost. For systems with , formula (A.8) could not be converged at all. We emphasize that formula (3.5) applies only when and are taken to be the same for different ’s. Additionally, they should be sufficiently large, so that analytical integration in the partition function applies. In practice, this can be achieved by gradually increasing and while monitoring at . The point at which this function becomes equal to the energy of the harmonic approximation indicates that the spring constants are strong enough. Here, is the number of degrees of freedom in the system. For the model where all particles are allowed to rotate and for the model with coupled rotations .
Integration of was carried out numerically. To counter a very strong decline of in the limit of , the following change of variables was performed:
| (A.9) |
The integral in equation (3.5) was transformed accordingly:
| (A.10) |
where parameters and were determined by fitting. The constant was set equal .
Both and the derivative of this function were used to compute the integral. The latter can be extracted directly from simulations through the following relationship:
The derivative of the integrand in equation (A.10) can be found as
Numerical integration was carried out by the Euler–Maclaurin method [22]. Initially, there were 15 non-overlapping segments considered with widths adjusted iteratively to achieve . This allowed us to make the numerical error almost uniform across the integration range. Each segment was integrated using the 2-point formula , where , and are the values of the integrand and its first derivative at the first point of the segment and and are the corresponding quantities at the second point of the segment. Each segment was then split evenly in two, yielding an intermediate point , and integration was repeated using the 3-point formula , where is the value of the function at and . Since the Euler–Maclaurin formula is accurate up to , the two estimates, and can be combined in a Romberg-style procedure to yield a better approximation for the integral , which is accurate up to terms . The error contained in this estimate can be approximated by , which is the error of the 3-point integration formula. If the error estimated in this way turned out to be higher than a pre-set target value, we continued to split the concerned segments until a desired accuracy was reached. A total of 33 integration points were generated in this manner. The grid was much denser for points corresponding to . The estimated integration error is less than 0.1% in relative terms. For the free energy difference, this translates into a 3% numerical error, which is about twice as low as the statistical error resulting from incomplete sampling.
References
- [1] van Gunsteren W. F., Dolenc J., Biochem. Soc. Trans., 2008, 36, 11, doi:10.1042/BST0360011.
- [2] Frenkel D., Smit B., Understanding Molecular Simulation, Academic Press, San Diego, 2002.
- [3] Panagiotopoulos A. Z., In: Observation, Prediction and Simulation of Phase Transitions in Complex Fluids, Vol. 460, Baus M., Rull L. F., Ryckaert J. P. (Eds.), Springer, Dordrecht, 1995, 463–501, doi:10.1007/978-94-011-0065-6_11.
- [4] Hukushima K., Nemoto K., J. Phys. Soc. Jpn., 1996, 65, No. 6, 1604, doi:10.1143/JPSJ.65.1604.
- [5] Swendsen R. H., Wang J. S., Phys. Rev. Lett., 1986, 57, No. 21, 2607, doi:10.1103/PhysRevLett.57.2607.
-
[6]
Tesi M. C., van Rensburg E. J. J., Orlandini E., Whittington S. G., J. Stat. Phys., 1996, 82, 155,
doi:10.1007/BF02189229. - [7] Fortini A., Dijkstra M., J. Phys.: Condens. Matter, 2006, 18, No. 28, L371, doi:10.1088/0953-8984/18/28/L02.
- [8] Noya E. G., Vega C., de Miguel E., J. Chem. Phys., 2008, 128, No. 15, 154507, doi:10.1063/1.2901172.
- [9] Allen M. P., Tildesley D. J., Computer Simulations of Liquids, Oxford University Press, Oxford, 1987.
- [10] Veerman J. A. C., Frenkel D., Phys. Rev. A, 1990, 41, 3237, doi:10.1103/PhysRevA.41.3237.
- [11] Bolhuis P., Frenkel D., J. Chem. Phys., 1997, 106, No. 2, 666, doi:10.1063/1.473404.
- [12] Rodgers J. M., Smit B., J. Chem. Theory Comput., 2012, 8, No. 2, 404, doi:10.1021/ct2007204.
- [13] Vieillard-Baron J., J. Chem. Phys., 1972, 56, No. 10, 4729, doi:10.1063/1.1676946.
- [14] Cuesta J. A., Frenkel D., Phys. Rev. A, 1990, 42, No. 4, 2126, doi:10.1103/PhysRevA.42.2126.
- [15] Bautista-Carbajal G., Odriozola G., J. Chem. Phys., 2014, 140, No. 20, 204502, doi:10.1063/1.4878411.
- [16] Xu W. S., Li Y. W., Sun Z. Y., An L. J., J. Chem. Phys., 2013, 139, No. 2, 024501, doi:10.1063/1.4812361.
- [17] Torrie G. M., Valleau J. P., J. Comput. Phys., 1977, 23, No. 2, 187, doi:10.1016/0021-9991(77)90121-8.
- [18] Frenkel D., Ladd A. J. C., J. Chem. Phys., 1984, 81, No. 7, 3188, doi:10.1063/1.448024.
-
[19]
Khanna V., Anwar J., Frenkel D., Doherty M. F., Peters B., J. Chem. Phys., 2021, 154, No. 16, 164509,
doi:10.1063/5.0044833. - [20] Ferrenberg A. M., Swendsen R. H., Phys. Rev. Lett., 1989, 63, No. 12, 1195, doi:10.1103/PhysRevLett.63.1195.
- [21] Kumar S., Bouzida D., Swendsen R. H., Kollman P. A., Rosenberg J. M., J. Comput. Chem., 1992, 13, No. 8, 1011, doi:10.1002/jcc.540130812.
- [22] DeVries P. L., A First Course in Computational Physics, John Wiley Sons, Inc., 1993.
Ukrainian \adddialect\l@ukrainian0 \l@ukrainian
Äî àëãîðèòìó ïðîâåäåííÿ Ìîíòå Êàðëî ñèìóëÿöié â êîìiðêàõ ìîäåëþâàííÿ ç ïîñòiéíèì îá’єìîì òà çìiííîþ ôîðìîþ À. Áàóìêåòíåð
Iíñòèòóò ôiçèêè êîíäåíñîâàíèõ ñèñòåì ÍÀÍ Óêðà¿íè, âóë. Ñâєíöiöüêîãî, 1, 79011, Ëüâiâ, Óêðà¿íà