Multilingual Molecular Representation Learning via Contrastive Pre-training
Abstract
Molecular representation learning plays an essential role in cheminformatics. Recently, language model-based approaches have gained popularity as an alternative to traditional expert-designed features to encode molecules. However, these approaches only utilize a single molecular language for representation learning. Motivated by the fact that a given molecule can be described using different languages such as Simplified Molecular Line Entry System (SMILES), the International Union of Pure and Applied Chemistry (IUPAC), and the IUPAC International Chemical Identifier (InChI), we propose a multilingual molecular embedding generation approach called MM-Deacon (multilingual molecular domain embedding analysis via contrastive learning). MM-Deacon is pre-trained using SMILES and IUPAC as two different languages on large-scale molecules. We evaluated the robustness of our method on seven molecular property prediction tasks from MoleculeNet benchmark, zero-shot cross-lingual retrieval, and a drug-drug interaction prediction task.
1 Introduction
Drug discovery process involves screening of millions of compounds in the early stages of drug design, which is time consuming and expensive. Computer-aided drug discovery can reduce the time and cost involved in this process via automating various cheminformatics tasks Kontogeorgis and Gani (2004); Xu et al. (2017); Winter et al. (2019).
Traditional methods to encode molecules such as fingerprint generation rely heavily on molecular fragment-level operations on top of molecule graph constructed by molecular atoms and bonds Burden (1989); Bender and Glen (2004); Vogt and Bajorath (2008); Muegge and Mukherjee (2016). An example of such methods is Morgan fingerprint, also known as Extended-Connectivity Fingerprint (ECFP) Morgan (1965); Rogers and Hahn (2010), where a fixed binary hash function is applied on each atom and its neighborhood. These kinds of approaches focus on local features, hence they may not capture global information.
In addition to molecule graph, a given molecule can also be described with different languages such as Simplified Molecular Line Entry System (SMILES), the International Union of Pure and Applied Chemistry (IUPAC), and the IUPAC International Chemical Identifier (InChI). Particularly, SMILES is widely used to represent molecule structures as ASCII strings Weininger (1988); Favre and Powell (2013) at an atom and bond level. IUPAC nomenclature, on the other hand, serves the purpose of systematically naming organic compounds by basic words that indicate the structure of the compound and prioritize on functional groups to facilitate communication Panico et al. (1993). Fig. 1 shows a comparison of SMILES and IUPAC characteristics for the same molecule. The SMILES string is created by traversing the molecule graph, where each letter in the SMILES string (such as C, F, N, O in Fig. 1) corresponds to an atom on the graph, and other characters represent positions and connectivity. However, IUPAC names are akin to a natural language, and morphemes in the IUPAC name (like fluoro, prop, en, yl in this example) often represent specific types of substructure on the molecule graph, which are also responsible for characteristic chemical reactions of molecules.
Advances in natural language processing (NLP) have been very promising for molecule embedding generation and molecular property prediction Xu et al. (2017); Gómez-Bombarelli et al. (2018); Samanta et al. (2020); Koge et al. (2021); Honda et al. (2019); Shrivastava and Kell (2021); Goh et al. (2017); Schwaller et al. (2019); Payne et al. (2020); Aumentado-Armstrong (2018). It is important to note that all of the methods mentioned above work with SMILES representation only. Therefore, the underlying chemical knowledge encoded in the embedding is restricted to a single language modality. Transformer models trained with self-supervised masked language modeling (MLM) loss Vaswani et al. (2017) in chemical domain Wang et al. (2019); Chithrananda et al. (2020); Elnaggar et al. (2020); Rong et al. (2020); Schwaller et al. (2021); Bagal et al. (2021) have also been used for molecular representation learning. However, pre-training objectives like MLM loss tend to impose task-specific bias on the final layers of Transformers Carlsson et al. (2020), limiting the generalization of the embeddings.
In recent years, contrastive learning has been successful in multimodal vision and language research Radford et al. (2021); Meyer et al. (2020); Shi et al. (2020); Cui et al. (2020); Chen et al. (2021); Alayrac et al. (2020); Akbari et al. (2021); Lee et al. (2020); Liu et al. (2020). Radford et al. (2021) used image-text pairs to learn scalable visual representations. Carlsson et al. (2020) showed the superiority of contrastive objectives in acquiring global (not fragment-level) semantic representations.

In light of these advances, we propose MM-Deacon (multilingual molecular domain embedding analysis via contrastive learning), a molecular representation learning algorithm built on SMILES and IUPAC joint training. Transformers are used as base encoders in MM-Deacon to encode SMILES and IUPAC, and embeddings from encoders are projected to a joint embedding space. Afterwards, a contrastive objective is used to push the embeddings of positive cross-lingual pairs (SMILES and IUPAC for the same molecule) closer together and the embeddings of negative cross-lingual pairs (SMILES and IUPAC for different molecules) farther apart. Here instead of using SMILES and IUPAC for sequence-to-sequence translation Rajan et al. (2021); Krasnov et al. (2021); Handsel et al. (2021), we obtain positive and negative SMILES-IUPAC pairs and contrast their embeddings at the global molecule level rather than the fragment level. Different molecule descriptors are thus integrated into the same joint embedding space, with mutual information maximized across distinct molecule languages.
We pre-train MM-Deacon on 10 million molecules chosen at random from the publicly available PubChem dataset Kim et al. (2016) and then use the pre-trained model for downstream tasks. Our main contributions are as follows:
-
•
We propose MM-Deacon, a novel approach for utilizing multiple molecular languages to generate molecule embeddings via contrastive learning.
-
•
To the best of our knowledge, we are the first to leverage mutual information shared across SMILES and IUPAC for molecule encoding.
-
•
We conduct extensive experiments on a variety of tasks, including molecular property prediction, cross-lingual molecule retrieval, and drug-drug interaction (DDI) prediction, and demonstrate that our approach outperforms baseline methods and existing state-of-the-art approaches.
2 Molecule pre-training
Deep learning tasks commonly face two challenges: first, dataset size is often limited, and second, annotations are scarce and expensive. A pre-training scheme can benefit downstream tasks by leveraging large-scale unlabeled or weakly labeled data. Such pre-training and fine-tuning frameworks have recently sparked much interest in the molecular domain Hu et al. (2019); Samanta et al. (2020); Chithrananda et al. (2020); Rong et al. (2020); Shrivastava and Kell (2021); Xue et al. (2021); Zhu et al. (2021); Wang et al. (2021); Liu et al. (2021). Existing pre-training methods can be divided into three categories based on the models used: pre-training with graph neural networks (GNNs), pre-training with language models, and pre-training with hybrid models.
Pre-training with GNNs. GNNs are a popular choice for molecule encoding that regard atoms as nodes and bonds as edges. Hu et al. (2019) pre-trained GNNs on 2 million molecules using both node-level and graph-level representations with attribute masking and structure prediction objectives. MolCLR Wang et al. (2021) used subgraph-level molecule data augmentation scheme to create positive and negative pairs and contrastive learning to distinguish positive from negative. GraphMVP Liu et al. (2021) was pre-trained on the consistency of 2D and 3D molecule graphs (3D graphs formed by adding atom spatial positions to 2D graphs) and contrastive objectives with GNNs.
Pre-training with language models. Language models are widely used to encode SMILES for molecular representation learning. Xu et al. (2017) reconstructed SMILES using encoder-decoder gated recurrent units (GRUs) with seq2seq loss, where embeddings in the latent space were used for downstream molecular property prediction. Chemberta Chithrananda et al. (2020) fed SMILES into Transformers, which were then optimized by MLM loss. FragNet Shrivastava and Kell (2021) used encoder-decoder Transformers to reconstruct SMILES and enforced extra supervision to the latent space with augmented SMILES and contrastive learning. X-Mol Xue et al. (2021) was pretrained by taking as input a pair of SMILES variants for the same molecule and generating one of the two input SMILES as output with Transformers on 1.1 billion molecules.
Pre-training with hybrid models. Different molecule data formats can be used collaboratively to enforce cross-modality alignment, resulting in the use of hybrid models. For example, DMP Zhu et al. (2021) was built on the consistency of SMILES and 2D molecule graphs, with SMILES encoded by Transformers and 2D molecule graphs encoded by GNNs.
Unlike other molecule pre-training methods, MM-Deacon is multilingually pre-trained with language models using pairwise SMILES and IUPAC. Compared with using molecule graphs with GNNs, IUPAC names encoded by language models bring in a rich amount of prior knowledge by basic words representing functional groups, without the need for sophisticated graph hyperparameter design.
3 Method

MM-Deacon is a deep neural network designed for SMILES-IUPAC joint learning with the goal of contrasting positive SMILES-IUPAC pairs from negative pairs and thus maximizing mutual information across different molecule languages. SMILES and IUPAC for the same molecule are regarded as positive pairs, while SMILES and IUPAC for different molecules are considered negative. Transformer encoders with multi-head self-attention layers are utilized to encode SMILES and IUPAC strings. Embeddings from the encoders are pooled globally and projected to the joint chemical embedding space. MM-Deacon is pre-trained on a dataset of 10 million molecules chosen at random from PubChem.
3.1 Tokenizer
We use a Byte-Pair Encoding (BPE) tokenizer for SMILES tokenization, as is shown by Chithrananda et al. (2020) that BPE performed better than regex-based tokenization for SMILES on downstream tasks. For IUPAC name tokenization, a rule-based regex Krasnov et al. (2021) that splits IUPAC strings based on suffixes, prefixes, trivial names, and so on is employed. The input sequence length statistics as well as the top 20 most frequent tokens in the SMILES and IUPAC corpora are displayed in Figs. 9 and 10 (Appendix A).
3.2 Model architecture
As illustrated in Fig. 2, MM-Deacon takes SMILES and IUPAC strings as the input to separate branches. The input text string is tokenized and embedded into a numeric matrix representation within each branch, and the order of the token list is preserved by a positional embedding . Then and are ingested by an encoder block that consists of 6 layers of Transformer encoder. A Transformer encoder has two sub-layers, a multi-head attention layer and a fully-connected feed-forward layer. Each sub-layer is followed by a residual connection and layer normalization to normalize input values for all neurons in the same layer Vaswani et al. (2017); Ba et al. (2016). The multi-head attention layer acquires long-dependency information by taking all positions into consideration. We then use a global average pooling layer to integrate features at all positions and a projection layer to project the integrated feature vector to the joint embedding space. Thus the final embedding of can be expressed as,
| (1) |
The maximum input token sequence length is set to 512. For each of the 6 Transformer encoder layers, we choose the number of self-attention heads as 12 and hidden size of 768. The projection layer projects the vector from length of 768 to 512 to make the representation more compact. Thus .
3.2.1 Contrastive loss
Our goal is to align pairs of language modalities in the joint embedding space by maximizing mutual information of positive pairs and distinguishing them from negative pairs. For this purpose, we use InfoNCE Oord et al. (2018); Alayrac et al. (2020); Radford et al. (2021) as the contrastive loss. We do not construct negative pairs manually. Instead, during training, we obtain negative pairs in minibatches. Using a minibatch of SMILES-IUPAC pairs from molecules as input, positive pairs and negative pairs can be generated within the correlation matrix of SMILES strings and IUPAC strings. More specifically, the only positive pair for -th SMILES is -th IUPAC, while the remaining IUPAC strings form negative pairs with -th SMILES. Therefore, the InfoNCE loss for -th SMILES is,
| (2) |
where and represent SMILES and IUPAC respectively. is the pairwise similarity function that employs cosine similarity in this work. is the temperature. Likewise, the loss function for -th IUPAC is,
| (3) |
As a result, the final loss function is as follows,
| (4) |
We pre-train MM-Deacon on 80 V100 GPUs for 10 epochs (15 hours in total) with a 16 batch size on each GPU using AdamW optimizer with a learning rate of . The temperature is set as 0.07 as in Oord et al. (2018).

3.3 Downstream stage
Knowledge gained during pre-training can be transferred to downstream tasks in different ways. Fig. 3 lists two situations that make use of pre-trained MM-Deacon in the downstream stage.
MM-Deacon fine-tuning: A task-specific classification/regression head can be attached to pre-trained MM-Deacon and the system as a whole can be tuned on downstream task datasets.
MM-Deacon fingerprint: Pre-trained MM-Deacon is frozen. An input molecule is embedded as MM-Deacon fingerprint for zero-shot explorations (such as clustering analysis and similarity retrieval) and supervised tasks with the help of an extra classifier.
4 Experiments
MM-Deacon was evaluated on seven molecular property prediction tasks from MoleculeNet benchmark Wu et al. (2018), zero-shot cross-lingual retrieval, and a drug-drug interaction (DDI) prediction task.
4.1 Molecular property prediction
MoleculeNet benchmark provides a unified framework for evaluating and comparing molecular machine learning methods on a variety of molecular property prediction tasks ranging from molecular quantum mechanics to physiological themes, and is widely acknowledged as the standard in the research community Hu et al. (2019); Chithrananda et al. (2020); Xue et al. (2021); Zhu et al. (2021); Wang et al. (2021); Liu et al. (2021). Four classification datasets and three regression datasets from the MoleculeNet benchmark were utilized to evaluate our approach.
Data. The blood-brain barrier penetration (BBBP), clinical trail toxicity (ClinTox), HIV replication inhibition (HIV), and side effect resource (SIDER) datasets are classification tasks in which molecule SMILES strings and their binary labels are provided in each task. Area Under Curve of the Receiver Operating Characteristic curve (ROC-AUC) is the performance metric in which the higher the value, the better the performance. For datasets with multiple tasks like SIDER, the averaged ROC-AUC across all tasks under the same dataset is reported. The fractions of train/val/test sets for each classification task are 0.8/0.1/0.1 with Scaffold split. Note that data split using molecule scaffolds (two-dimensional structural frameworks) results in more structurally distinct train/val/test sets, making it more challenging than random split Wu et al. (2018). The water solubility data (ESOL), free solvation (FreeSolv), and experimental results of octabol/water distribution coefficient (Lipophilicity) datasets are all regression tasks to predict numeric labels given molecule SMILES strings. Root Mean Square Error (RMSE) is used as the evaluation metric in which the lower the value, the better the performance. As recommended by MoleculeNet, random split that divides each dataset into 0.8/0.1/0.1 for train/val/test sets is employed. The results on validation set are used to select the best model. To maintain consistency with MoleculeNet, we ran each task three times, each time with a different data split seed, to obtain the mean and standard deviation (std) of the metric. Details of each dataset such as the number of tasks and molecules it contains are displayed in Table 1.
| Dataset | # Molecules | # Tasks | Split | Metric |
|---|---|---|---|---|
| BBBP | 2039 | 1 | Scaffold | ROC-AUC |
| ClinTox | 1478 | 2 | Scaffold | ROC-AUC |
| HIV | 41127 | 1 | Scaffold | ROC-AUC |
| SIDER | 1427 | 27 | Scaffold | ROC-AUC |
| ESOL | 1128 | 1 | Random | RMSE |
| FreeSolv | 642 | 1 | Random | RMSE |
| Lipophilicity | 4200 | 1 | Random | RMSE |
Model. We utilized the model shown in Fig. 3(a) in which a linear layer serving as the task-specific head was added to pre-trained MM-Deacon SMILES branch for fine-tuning (IUPAC branch was removed). Cross-entropy loss was employed for classification tasks and MSE loss was employed for regression tasks. Hyperparameter tuning was performed using grid search with possible choices listed in Table 5 (Appendix B). Each task was optimized individually.
| Method | BBBP | ClinTox | HIV | SIDER | ESOL | FreeSolv | Lipophilicity |
| RF | 71.40.0 | 71.35.6 | 78.10.6 | 68.40.9 | 1.070.19 | 2.030.22 | 0.8760.040 |
| KernelSVM | 72.90.0 | 66.99.2 | 79.20.0 | 68.21.3 | - | - | - |
| Multitask | 68.80.5 | 77.85.5 | 69.83.7 | 66.62.6 | 1.120.15 | 1.870.07 | 0.8590.013 |
| GC | 69.00.9 | 80.74.7 | 76.31.6 | 63.81.2 | 0.970.01 | 1.400.16 | 0.6550.036 |
| Weave | 67.11.4 | 83.23.7 | 70.33.9 | 58.12.7 | 0.610.07 | 1.220.28 | 0.7150.035 |
| MPNN | - | - | - | - | 0.580.03 | 1.150.12 | 0.7190.031 |
| Hu et al. (2019) | 70.81.5 | 78.92.4 | 80.20.9 | 65.20.9 | - | - | - |
| MolCLR Wang et al. (2021) | 73.60.5 | 93.21.7 | 80.61.1 | 68.01.1 | - | - | - |
| DMP Zhu et al. (2021) | 78.10.5 | 95.00.5 | 81.00.7 | 69.20.7 | - | - | - |
| X-Mol Xue et al. (2021) | 96.2N/A | 98.4N/A | 79.8N/A | - | 0.578N/A | 1.108N/A | 0.596N/A |
| GraphMVP Liu et al. (2021) | 72.41.6 | 77.54.2 | 77.01.2 | 63.91.2 | 1.029N/A | - | 0.681N/A |
| MLM-[CLS] | 70.64.5 | 93.20.1 | 77.90.2 | 64.81.3 | 0.6400.023 | 1.210.046 | 0.8040.037 |
| MM-Deacon | 78.50.4 | 99.50.3 | 80.10.5 | 69.30.5 | 0.5650.014 | 0.9260.013 | 0.6500.021 |
Results. Table 2 shows the mean and std results for each dataset. The first half of the table displays results imported from MoleculeNet Wu et al. (2018), while the second section shows the results from MM-Deacon and other state-of-the-art molecular pre-training and fine-tuning approaches. MLM-[CLS] denotes our implementation of a Chemberta Chithrananda et al. (2020) variant that uses the same Transformer settings as MM-Deacon SMILES branch, pre-trained with MLM loss on 10M molecules, and fine-tuned through [CLS] token with the same downstream setting as MM-Deacon. MM-Deacon exceeds the performance of traditional machine learning methods like random forest (RF) and task-specific GNNs reported in MoleculeNet work by a significant margin for most of the tasks. When compared to other pre-training based approaches, MM-Deacon outperforms the existing state-of-the-art approaches in four of the seven datasets and is comparable in the remaining three, with major improvements on ClinTox and FreeSolv.
All pre-training based methods were pre-trained on millions of molecules, with the exception of GraphMVP, which was pre-trained on 50K molecules. The requirement that molecules have both 2D and 3D structure information available at the same time to be qualified has limited the scalability of GraphMVP. MM-Deacon and MLM-CLS both used 6 layers of Transformer blocks to process SMILES. For each task, MM-Deacon, which was pre-trained with both SMILES and IUPAC, outscored MLM-CLS, which was pre-trained with SMILES only. MM-Deacon and DMP performed comparably on the four classification tasks, while DMP used 12 layers of Transformer blocks for SMILES and a 12-layer GNN to encode a molecule 2D graph, which is nearly twice the size of MM-Deacon model.
Moreover, we found that BBBP test set is significantly more challenging than the validation set, which is consistent with the results published in the MoleculeNet paper Wu et al. (2018). The substantially high accuracy X-Mol achieved on the BBBP dataset could be due to either the 1.1 billion molecules they utilized for pre-training or a different dataset division approach they employed.
4.2 Zero-shot cross-lingual retrieval
In addition to conducting fine-tuning on supervised tasks like molecular property prediction, pre-trained MM-Deacon can be employed directly in large-scale zero-shot analysis. Zero-shot cross-lingual retrieval operates on top of MM-Deacon fingerprint generated by pre-trained MM-Deacon given molecule SMILES or IUPAC as input. This task enables the retrieval of similar molecules across languages without the need for translation, and it can also be used to evaluate the learned agreement in the joint embedding space between SMILES and IUPAC representations.
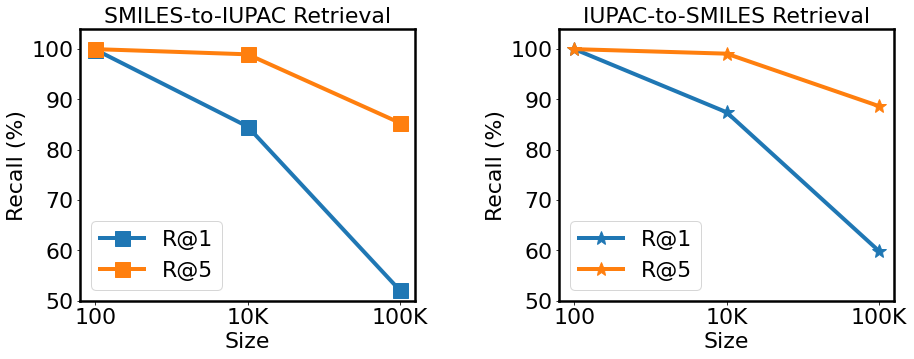
Data. 100K molecules were randomly chosen from PubChem dataset after excluding the 10 million molecules used for MM-Deacon pre-training. SMILES and IUPAC strings are provided for each molecule. We used average recall at K (R@1 and R@5) to measure the percentage of the ground truth that appears in the top K retrieved molecules.
Model. Pre-trained MM-Deacon was used for MM-Deacon fingerprint generation, as shown in Fig. 3(b). As a result, each SMILES and IUPAC string was encoded as MM-Deacon SMILES fingerprint and IUPAC fingerprint respectively. Cosine similarity between a query and molecules in the search candidates was used to determine the ranking.
Results. Fig. 4 shows the outcomes of SMILES-to-IUPAC and IUPAC-to-SMILES retrieval in terms of recall. We not only performed retrieval directly on the entire 100K molecules, but also reported the results on smaller groups of molecules (100, 10K) to get a more thorough picture of the retrieval performance. MM-Deacon gets a R@5 above 85% for both types of cross-lingual retrieval even while executing retrieval on 100K molecules. Moreover, Figs. 5 and 6 show an example of SMILES-to-IUPAC retrieval and an example of IUPAC-to-SMILES retrieval respectively.


Additional retrieval examples for scenarios where the performance is difficult to be quantified, such as retrieval queried by a free combination of tokens and unilingual retrieval are included in Appendix C.
4.3 DDI prediction
. Method AUC AUPR Precision Recall Neighbor recommender*‡ 0.936 0.759 0.617 0.765 Random walk*‡ 0.936 0.758 0.763 0.616 Ensemble method (L1)* 0.957 0.807 0.785 0.670 Ensemble method (L2)* 0.956 0.806 0.783 0.665 DPDDI Feng et al. (2020) 0.956 0.907 0.754 0.810 MLM-[CLS]† 0.943 0.901 0.784 0.813 MM-Deacon (SMILES)† 0.946 0.911 0.805 0.823 MM-Deacon (IUPAC)† 0.947 0.913 0.834 0.797 MM-Deacon (concat)† 0.950 0.918 0.819 0.824
The effectiveness of combining MM-Deacon fingerprints with a task-specific classifier for supervised learning was tested on a DDI prediction task. The objective of this task is to predict whether or not any two given drugs have an interaction.
Data. The DDI dataset Zhang et al. (2017) used here includes 548 drugs, with 48,584 known interactions, and 101,294 non-interactions (may contain undiscovered interactions at the time the dataset was created). We obtained the SMILES and IUPAC names for each drug from PubChem. Stratified 5-fold cross-validation with drug combination split was utilized. The evaluation metrics are Area Under the ROC Curve (AUC), Area Under the Precision-Recall Curve (AUPR), precision, and recall, with AUPR serving as the primary metric Zhang et al. (2017).
Model. MM-Deacon fingerprints of paired drugs are concatenated and fed into a multi-layer perceptron (MLP) network implemented by scikit-learn Pedregosa et al. (2011) for binary classification. Three different types of fingerprints are used for MM-Deacon: SMILES, IUPAC, and concatenated SMILES and IUPAC fingerprints. The MLP has one hidden layer with 200 neurons. ReLU activation and a learning rate of are used.
Results. As shown in Table 3, MM-Deacon outperforms other methods in terms of AUPR, precision and recall, with the maximum AUPR obtained when SMILES and IUPAC fingerprints were concatenated as input feature set. Ensemble models Zhang et al. (2017) included extra bioactivity related features in addition to drug structural properties. DPDDI Feng et al. (2020) encoded molecule graph with GNNs, from which latent features were concatenated for pairs of drugs and ingested into a deep neural network.
Table 4 shows the top 20 most potential interactions predicted by MM-Deacon (concat) in the non-interaction set (false positives), 13 out of which are confirmed as true positives by DrugBank111https://go.drugbank.com/drug-interaction-checker. While, the number is 7/20 for ensemble models Zhang et al. (2017).
| Rank | Drug 1 ID (Name) | Drug 2 ID (Name) |
|---|---|---|
| 1 | DB00722 (Lisinopril) | DB00915 (Amantadine) |
| 2 | DB00213 (Pantoprazole) | DB00310 (Chlorthalidone) |
| 3 | DB00776 (Oxcarbazepine) | DB00433 (Prochlorperazine) |
| 4 | DB00481 (Raloxifene) | DB00501 (Cimetidine) |
| 5 | DB01193 (Acebutolol) | DB00264 (Metoprolol) |
| 6 | DB00250 (Dapsone) | DB00230 (Pregabalin) |
| 7 | DB00415 (Ampicillin) | DB01112 (Cefuroxime) |
| 8 | DB00582 (Voriconazole) | DB01136 (Carvedilol) |
| 9 | DB01079 (Tegaserod) | DB00795 (Sulfasalazine) |
| 10 | DB01233 (Metoclopramide) | DB00820 (Tadalafil) |
| 11 | DB00213 (Pantoprazole) | DB00513 (Aminocaproic Acid) |
| 12 | DB01195 (Flecainide) | DB00584 (Enalapril) |
| 13 | DB00758 (Clopidogrel) | DB01589 (Quazepam) |
| 14 | DB01136 (Carvedilol) | DB00989 (Rivastigmine) |
| 15 | DB00586 (Diclofenac) | DB01149 (Nefazodone) |
| 16 | DB00407 (Ardeparin) | DB00538 (Gadoversetamide) |
| 17 | DB00203 (Sildenafil) | DB00603 (Medroxyprogesterone Acetate) |
| 18 | DB00601 (Linezolid) | DB01112 (Cefuroxime) |
| 19 | DB00275 (Olmesartan) | DB00806 (Pentoxifylline) |
| 20 | DB00231 (Temazepam) | DB00930 (Colesevelam) |
5 Discussions
After being pre-trained on 10 million molecules, MM-Deacon showed outstanding knowledge transfer capabilities to various downstream scenarios (Fig. 3) where a pre-trained model could be used. The competitive performance on seven molecular property prediction tasks from MoleculeNet benchmark demonstrated the effectiveness of the pre-trained MM-Deacon when adopting a network fine-tuning scheme as shown in Fig. 3(a). The evaluation results of zero-shot cross-lingual retrieval further revealed that MM-Deacon SMILES and IUPAC fingerprints shared a substantial amount of mutual information, implying that an IUPAC name can be used directly without first being translated to SMILES format as chemists have done in the past. The DDI prediction task showed that MM-Deacon also allows directly using embeddings in the joint cross-modal space as molecular fingerprints for downstream prediction tasks, which is a widely used strategy in cheminformatics.
MM-Deacon profited from the alignment of two molecule languages with distinct forms of nomenclatures, as opposed to the baseline MLM-[CLS] model, which was pre-trained on SMILES representation only. Furthermore, we looked at molecule-level and token-level alignments of MM-Deacon to untangle the outcome of cross-lingual contrastive learning.

5.1 Molecule-level alignment
We used centered kernel alignment (CKA) Kornblith et al. (2019) with RBF kernel to compare representations between different layers. In Fig. 7(a), the representations of 6 Transformer layers and the final projection layer were compared between MM-Deacon SMILES and IUPAC branches, where the representations differ in shallow layers, while reach a high level of alignment in deeper layers. In Fig. 7(b), both the MM-Deacon SMILES branch and MLM-[CLS] model take SMILES as the input, therefore the shallow layers have a high alignment score, while the representation varies as the network grows deeper. Fig. 7 shows that MM-Deacon aligned SMILES and IUPAC representations effectively, and that molecular representations trained with SMILES and IUPAC differs from representations trained only on SMILES.
5.2 Token-level alignment
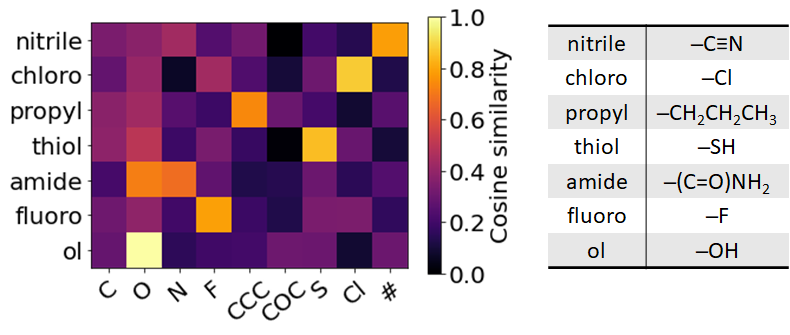
The cosine similarity matrix of MM-Deacon fingerprints between tokens from the IUPAC corpus and tokens from the SMILES corpus is shown in Fig. 8. The table in Fig. 8 lists IUPAC tokens expressed in SMILES language, and the heat map demonstrates that there exists a good token-level alignment between SMILES and IUPAC.
6 Conclusion
In this study, we proposed a novel method for multilingual molecular representation learning that combines mutual information from SMILES-IUPAC joint training with a self-supervised contrastive loss. We evaluated our approach for molecular property prediction, zero-shot cross-lingual retrieval, and DDI prediction. Our results demonstrate that the self-supervised multilingual contrastive learning framework holds enormous possibilities for chemical domain exploration and drug discovery. In future work, we plan to scale MM-Deacon pre-training to larger dataset sizes, as well as investigate the applicability of MM-Deacon to other types of molecule languages.
Acknowledgements
We would like to thank Min Xiao and Brandon Smock for some insightful discussions.
References
- Akbari et al. (2021) Hassan Akbari, Linagzhe Yuan, Rui Qian, Wei-Hong Chuang, Shih-Fu Chang, Yin Cui, and Boqing Gong. 2021. Vatt: Transformers for multimodal self-supervised learning from raw video, audio and text. arXiv preprint arXiv:2104.11178.
- Alayrac et al. (2020) Jean-Baptiste Alayrac, Adria Recasens, Rosalia Schneider, Relja Arandjelovic, Jason Ramapuram, Jeffrey De Fauw, Lucas Smaira, Sander Dieleman, and Andrew Zisserman. 2020. Self-supervised multimodal versatile networks. NeurIPS, 2(6):7.
- Aumentado-Armstrong (2018) Tristan Aumentado-Armstrong. 2018. Latent molecular optimization for targeted therapeutic design. arXiv preprint arXiv:1809.02032.
- Ba et al. (2016) Jimmy Lei Ba, Jamie Ryan Kiros, and Geoffrey E Hinton. 2016. Layer normalization. arXiv preprint arXiv:1607.06450.
- Bagal et al. (2021) Viraj Bagal, Rishal Aggarwal, PK Vinod, and U Deva Priyakumar. 2021. Liggpt: Molecular generation using a transformer-decoder model.
- Bender and Glen (2004) Andreas Bender and Robert C Glen. 2004. Molecular similarity: a key technique in molecular informatics. Organic & biomolecular chemistry, 2(22):3204–3218.
- Burden (1989) Frank R Burden. 1989. Molecular identification number for substructure searches. Journal of Chemical Information and Computer Sciences, 29(3):225–227.
- Carlsson et al. (2020) Fredrik Carlsson, Amaru Cuba Gyllensten, Evangelia Gogoulou, Erik Ylipää Hellqvist, and Magnus Sahlgren. 2020. Semantic re-tuning with contrastive tension. In International Conference on Learning Representations.
- Chen et al. (2021) Brian Chen, Andrew Rouditchenko, Kevin Duarte, Hilde Kuehne, Samuel Thomas, Angie Boggust, Rameswar Panda, Brian Kingsbury, Rogerio Feris, David Harwath, et al. 2021. Multimodal clustering networks for self-supervised learning from unlabeled videos. arXiv preprint arXiv:2104.12671.
- Chithrananda et al. (2020) Seyone Chithrananda, Gabriel Grand, and Bharath Ramsundar. 2020. Chemberta: Large-scale self-supervised pretraining for molecular property prediction. arXiv preprint arXiv:2010.09885.
- Cui et al. (2020) Wanyun Cui, Guangyu Zheng, and Wei Wang. 2020. Unsupervised natural language inference via decoupled multimodal contrastive learning. arXiv preprint arXiv:2010.08200.
- Elnaggar et al. (2020) Ahmed Elnaggar, Michael Heinzinger, Christian Dallago, Ghalia Rihawi, Yu Wang, Llion Jones, Tom Gibbs, Tamas Feher, Christoph Angerer, Martin Steinegger, et al. 2020. Prottrans: towards cracking the language of life’s code through self-supervised deep learning and high performance computing. arXiv preprint arXiv:2007.06225.
- Favre and Powell (2013) Henri A Favre and Warren H Powell. 2013. Nomenclature of organic chemistry: IUPAC recommendations and preferred names 2013. Royal Society of Chemistry.
- Feng et al. (2020) Yue-Hua Feng, Shao-Wu Zhang, and Jian-Yu Shi. 2020. Dpddi: a deep predictor for drug-drug interactions. BMC bioinformatics, 21(1):1–15.
- Goh et al. (2017) Garrett B Goh, Nathan O Hodas, Charles Siegel, and Abhinav Vishnu. 2017. Smiles2vec: An interpretable general-purpose deep neural network for predicting chemical properties. arXiv preprint arXiv:1712.02034.
- Gómez-Bombarelli et al. (2018) Rafael Gómez-Bombarelli, Jennifer N Wei, David Duvenaud, José Miguel Hernández-Lobato, Benjamín Sánchez-Lengeling, Dennis Sheberla, Jorge Aguilera-Iparraguirre, Timothy D Hirzel, Ryan P Adams, and Alán Aspuru-Guzik. 2018. Automatic chemical design using a data-driven continuous representation of molecules. ACS central science, 4(2):268–276.
- Handsel et al. (2021) Jennifer Handsel, Brian Matthews, Nicola Knight, and Simon Coles. 2021. Translating the molecules: adapting neural machine translation to predict iupac names from a chemical identifier.
- Honda et al. (2019) Shion Honda, Shoi Shi, and Hiroki R Ueda. 2019. Smiles transformer: Pre-trained molecular fingerprint for low data drug discovery. arXiv preprint arXiv:1911.04738.
- Hu et al. (2019) Weihua Hu, Bowen Liu, Joseph Gomes, Marinka Zitnik, Percy Liang, Vijay Pande, and Jure Leskovec. 2019. Strategies for pre-training graph neural networks. arXiv preprint arXiv:1905.12265.
- Kim et al. (2016) Sunghwan Kim, Paul A Thiessen, Evan E Bolton, Jie Chen, Gang Fu, Asta Gindulyte, Lianyi Han, Jane He, Siqian He, Benjamin A Shoemaker, et al. 2016. PubChem substance and compound databases. Nucleic acids research, 44(D1):D1202–D1213.
- Koge et al. (2021) Daiki Koge, Naoaki Ono, Ming Huang, Md Altaf-Ul-Amin, and Shigehiko Kanaya. 2021. Embedding of molecular structure using molecular hypergraph variational autoencoder with metric learning. Molecular informatics, 40(2):2000203.
- Kontogeorgis and Gani (2004) Georgios M Kontogeorgis and Rafiqul Gani. 2004. Computer Aided Property Estimation for Process and Product Design: Computers Aided Chemical Engineering. Elsevier.
- Kornblith et al. (2019) Simon Kornblith, Mohammad Norouzi, Honglak Lee, and Geoffrey Hinton. 2019. Similarity of neural network representations revisited. In International Conference on Machine Learning, pages 3519–3529. PMLR.
- Krasnov et al. (2021) Lev Krasnov, Ivan Khokhlov, Maxim Fedorov, and Sergey Sosnin. 2021. Struct2iupac–transformer-based artificial neural network for the conversion between chemical notations.
- Landrum (2013) Greg Landrum. 2013. Rdkit: A software suite for cheminformatics, computational chemistry, and predictive modeling.
- Lee et al. (2020) Sangho Lee, Youngjae Yu, Gunhee Kim, Thomas Breuel, Jan Kautz, and Yale Song. 2020. Parameter efficient multimodal transformers for video representation learning. arXiv preprint arXiv:2012.04124.
- Liu et al. (2021) Shengchao Liu, Hanchen Wang, Weiyang Liu, Joan Lasenby, Hongyu Guo, and Jian Tang. 2021. Pre-training molecular graph representation with 3d geometry. arXiv preprint arXiv:2110.07728.
- Liu et al. (2020) Yunze Liu, Li Yi, Shanghang Zhang, Qingnan Fan, Thomas Funkhouser, and Hao Dong. 2020. P4contrast: Contrastive learning with pairs of point-pixel pairs for rgb-d scene understanding. arXiv preprint arXiv:2012.13089.
- McInnes et al. (2018) Leland McInnes, John Healy, and James Melville. 2018. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:1802.03426.
- Meyer et al. (2020) Johannes Meyer, Andreas Eitel, Thomas Brox, and Wolfram Burgard. 2020. Improving unimodal object recognition with multimodal contrastive learning. In 2020 IEEE/RSJ International Conference on Intelligent Robots and Systems (IROS), pages 5656–5663. IEEE.
- Morgan (1965) Harry L Morgan. 1965. The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. Journal of Chemical Documentation, 5(2):107–113.
- Muegge and Mukherjee (2016) Ingo Muegge and Prasenjit Mukherjee. 2016. An overview of molecular fingerprint similarity search in virtual screening. Expert opinion on drug discovery, 11(2):137–148.
- Oord et al. (2018) Aaron van den Oord, Yazhe Li, and Oriol Vinyals. 2018. Representation learning with contrastive predictive coding. arXiv preprint arXiv:1807.03748.
- Panico et al. (1993) R Panico, WH Powell, and Jean-Claude Richer. 1993. A guide to IUPAC Nomenclature of Organic Compounds, volume 2. Blackwell Scientific Publications, Oxford.
- Payne et al. (2020) Josh Payne, Mario Srouji, Dian Ang Yap, and Vineet Kosaraju. 2020. Bert learns (and teaches) chemistry. arXiv preprint arXiv:2007.16012.
- Pedregosa et al. (2011) F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot, and E. Duchesnay. 2011. Scikit-learn: Machine learning in Python. Journal of Machine Learning Research, 12:2825–2830.
- Radford et al. (2021) Alec Radford, Jong Wook Kim, Chris Hallacy, Aditya Ramesh, Gabriel Goh, Sandhini Agarwal, Girish Sastry, Amanda Askell, Pamela Mishkin, Jack Clark, et al. 2021. Learning transferable visual models from natural language supervision. arXiv preprint arXiv:2103.00020.
- Rajan et al. (2021) Kohulan Rajan, Achim Zielesny, and Christoph Steinbeck. 2021. Stout: Smiles to iupac names using neural machine translation. Journal of Cheminformatics, 13(1):1–14.
- Rogers and Hahn (2010) David Rogers and Mathew Hahn. 2010. Extended-connectivity fingerprints. Journal of chemical information and modeling, 50(5):742–754.
- Rong et al. (2020) Yu Rong, Yatao Bian, Tingyang Xu, Weiyang Xie, Ying Wei, Wenbing Huang, and Junzhou Huang. 2020. Self-supervised graph transformer on large-scale molecular data. arXiv preprint arXiv:2007.02835.
- Samanta et al. (2020) Soumitra Samanta, Steve O’Hagan, Neil Swainston, Timothy J Roberts, and Douglas B Kell. 2020. Vae-sim: a novel molecular similarity measure based on a variational autoencoder. Molecules, 25(15):3446.
- Schwaller et al. (2019) Philippe Schwaller, Teodoro Laino, Théophile Gaudin, Peter Bolgar, Christopher A Hunter, Costas Bekas, and Alpha A Lee. 2019. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS central science, 5(9):1572–1583.
- Schwaller et al. (2021) Philippe Schwaller, Daniel Probst, Alain C Vaucher, Vishnu H Nair, David Kreutter, Teodoro Laino, and Jean-Louis Reymond. 2021. Mapping the space of chemical reactions using attention-based neural networks. Nature Machine Intelligence, 3(2):144–152.
- Shi et al. (2020) Lei Shi, Kai Shuang, Shijie Geng, Peng Su, Zhengkai Jiang, Peng Gao, Zuohui Fu, Gerard de Melo, and Sen Su. 2020. Contrastive visual-linguistic pretraining. arXiv preprint arXiv:2007.13135.
- Shrivastava and Kell (2021) Aditya Divyakant Shrivastava and Douglas B Kell. 2021. Fragnet, a contrastive learning-based transformer model for clustering, interpreting, visualizing, and navigating chemical space. Molecules, 26(7):2065.
- Vaswani et al. (2017) Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N Gomez, Łukasz Kaiser, and Illia Polosukhin. 2017. Attention is all you need. In Advances in neural information processing systems, pages 5998–6008.
- Vogt and Bajorath (2008) Martin Vogt and Jürgen Bajorath. 2008. Bayesian screening for active compounds in high-dimensional chemical spaces combining property descriptors and molecular fingerprints. Chemical biology & drug design, 71(1):8–14.
- Wang et al. (2019) Sheng Wang, Yuzhi Guo, Yuhong Wang, Hongmao Sun, and Junzhou Huang. 2019. Smiles-bert: large scale unsupervised pre-training for molecular property prediction. In Proceedings of the 10th ACM international conference on bioinformatics, computational biology and health informatics, pages 429–436.
- Wang et al. (2021) Yuyang Wang, Jianren Wang, Zhonglin Cao, and Amir Barati Farimani. 2021. Molclr: Molecular contrastive learning of representations via graph neural networks. arXiv preprint arXiv:2102.10056.
- Weininger (1988) David Weininger. 1988. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of chemical information and computer sciences, 28(1):31–36.
- Winter et al. (2019) Robin Winter, Floriane Montanari, Frank Noé, and Djork-Arné Clevert. 2019. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chemical science, 10(6):1692–1701.
- Wu et al. (2018) Zhenqin Wu, Bharath Ramsundar, Evan N Feinberg, Joseph Gomes, Caleb Geniesse, Aneesh S Pappu, Karl Leswing, and Vijay Pande. 2018. Moleculenet: a benchmark for molecular machine learning. Chemical science, 9(2):513–530.
- Xu et al. (2017) Zheng Xu, Sheng Wang, Feiyun Zhu, and Junzhou Huang. 2017. Seq2seq fingerprint: An unsupervised deep molecular embedding for drug discovery. In Proceedings of the 8th ACM international conference on bioinformatics, computational biology, and health informatics, pages 285–294.
- Xue et al. (2021) Dongyu Xue, Han Zhang, Dongling Xiao, Yukang Gong, Guohui Chuai, Yu Sun, Hao Tian, Hua Wu, Yukun Li, and Qi Liu. 2021. X-mol: large-scale pre-training for molecular understanding and diverse molecular analysis. bioRxiv, pages 2020–12.
- Zhang et al. (2017) Wen Zhang, Yanlin Chen, Feng Liu, Fei Luo, Gang Tian, and Xiaohong Li. 2017. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC bioinformatics, 18(1):1–12.
- Zhu et al. (2021) Jinhua Zhu, Yingce Xia, Tao Qin, Wengang Zhou, Houqiang Li, and Tie-Yan Liu. 2021. Dual-view molecule pre-training. arXiv preprint arXiv:2106.10234.
Appendix A Data statistics
The distributions of SMILES and IUPAC sequence lengths in the training set are shown in Fig. 9 in log scale for the y axis.

Fig. 10 displays the top 20 most frequent alphabetic tokens in SMILES and IUPAC corpora. Token C, which simply denotes a carbon atom, appears nearly 20% of the time in SMILES language. On the other hand, the frequency of IUPAC tokens is quite evenly distributed, with the prefixes methyl and phenyl as well as the suffix yl from the alkyl functional group being the top 3 most common tokens.

Appendix B Hyperparameter tuning
Table 5 lists the search space of hyperparameter tuning for seven molecular property prediction tasks from MoleculeNet benchmark for MM-Deacon and MLM-[CLS]. We employed grid search to find the best hyperparameters. Each task was optimized individually.
. Parameter Choices Learning rate {1e-6, 5e-6, 1e-5, 5e-5, 1e-4} Batch size {2, 4, 8, 12, 24} Epochs 20 Early termination Bandit policy
Appendix C Extra zero-shot retrieval examples
An example of cross-lingual retrieval using a free-form IUPAC query is shown in Fig. 11. The IUPAC query thiolamide, a combination of tokens thiol and amide, does not exist in the IUPAC corpus (is not a substring of any IUPAC name). When searching on top of MM-Deacon fingerprints, all of the retrieved molecules have the features of atom S, N and C=O. That is, the semantic meaning of the query is captured.

In addition to cross-lingual retrieval, unilingual similarity retrieval is also supported, while its performance is difficult to be quantified. Figs. 12 and 13 show an example of SMILES-to-SMILES retrieval and IUPAC-to-IUPAC retrieval respectively using MM-Deacon fingerprints.

