Increase of barium ion-trap lifetime via photodissociation
Abstract
The lifetime of Ba+ ions confined in a Paul trap is found, under typical conditions, to be limited by chemical reactions with residual background gas. An integrated ion trap and time-of-flight mass spectrometer are used to analyze the reactions of the trapped Ba+ ions with three common gases in an ultrahigh vacuum system (H2, CO2 and H2O). It is found that the products of these reactions can all be photodissociated by a single ultraviolet laser at 225 nm, thereby allowing the recovery of the Ba+ ions and leading to an increase of the effective trap lifetime. For a Coulomb crystal, the lifetime increased from roughly 6 hours to 2 days at room temperature. It is suggested that higher enhancement factors are possible in systems with stronger traps. In addition, photodissociation wavelengths for other common trapped ion systems are provided.
To date, the highest-fidelity quantum operations and the largest ratio of qubit coherence time to gate time has been achieved in trapped ion systems Bruzewicz et al. (2019a); Christensen et al. (2020); Ballance et al. (2016); Srinivas et al. ; Gaebler et al. (2016). As such, ion-based qubits are a leading candidate for the construction of a large-scale quantum information platform, with major efforts underway using integrated photonics and microfabricated traps Bruzewicz et al. (2019b); Mehta et al. (2020). However, as these systems are scaled to a larger number of ions, the finite lifetime of ions in the trap, , leads to probability for loss of an ion , where is the number of ions in the register and is the time required for the entire quantum operation. While loss of an ion is generally detrimental to the operation at hand, it also triggers a time-consuming reloading operation. Further, the reloading process can often cause a number of deleterious effects, including unwanted charging of the trap electrodes. Several techniques have been developed to mitigate these problems, including loading from a magneto-optical trap Bruzewicz et al. (2016) and placing the ion trap in a cryogenic environment Pagano et al. (2018); Brandl et al. (2016); Leopold et al. (2019), where the residual background gas, which is typically assumed to be responsible for ion loss, is greatly reduced.
Here, we study the trap-loss process for Ba+ ions and find that ion loss is dominated by chemical reactions with residual background gas which produce molecular ions that, at high-enough trapping potential and with efficient sympathetic cooling, do not leave the ion trap. Building on the work of Ref. Sawyer et al. (2015); Chen et al. (2011) and guided by available spectroscopic data and ab initio molecular structure calculations, we demonstrate a simple technique for recovering the atomic ion qubit from the product molecular ion. Specifically, we employ photodissociation with a single laser that is capable of dissociating all common molecular ion products and increases the observed trapped-ion lifetime by a factor of in an ion chain and 7.6(8) for an ion crystal— here () denotes one standard error. The enhancement appears to be limited by the inability of the current ion trap to capture all of the recoiling product molecular ions. Systems with higher secular frequency Guggemos et al. (2015) will likely realize larger enhancement factors.
Besides reactive loss, micromotion interruption due to background gas represents another possible loss mechanism for ions. The time-varying nature of the ion trap confinement provides mechanisms for energy to be coupled from the radio-frequency (rf) electric potential that confines the ions into the ion motion during a collision with background gas, as well as with other ions. Refs. Chen et al. (2014, 2013); Schowalter et al. (2016); Cetina et al. (2012) analyzed this phenomenon and found that it is possible to achieve ion temperatures several times that of the background gas through such collisions. Therefore, elastic collisions with the background gas and then further collisions between the ions, can lead to ion loss.
To investigate the role of micromotion-interruption-induced loss, 138Ba+ are trapped and laser-cooled in a linear Paul trap with field radius mm, driven with an rf frequency at MHz and a peak-to-peak amplitude of 320 V, leading to a radial secular frequency of kHz. Axial confinement is provided by two DC electrodes spaced by 20 mm. The ions are detected either via imaging their laser-cooling-induced fluorescence through an objective with numerical aperture of 0.23 or by an integrated time-of-flight mass spectrometer— further details are provided in Ref. Mills (2020).
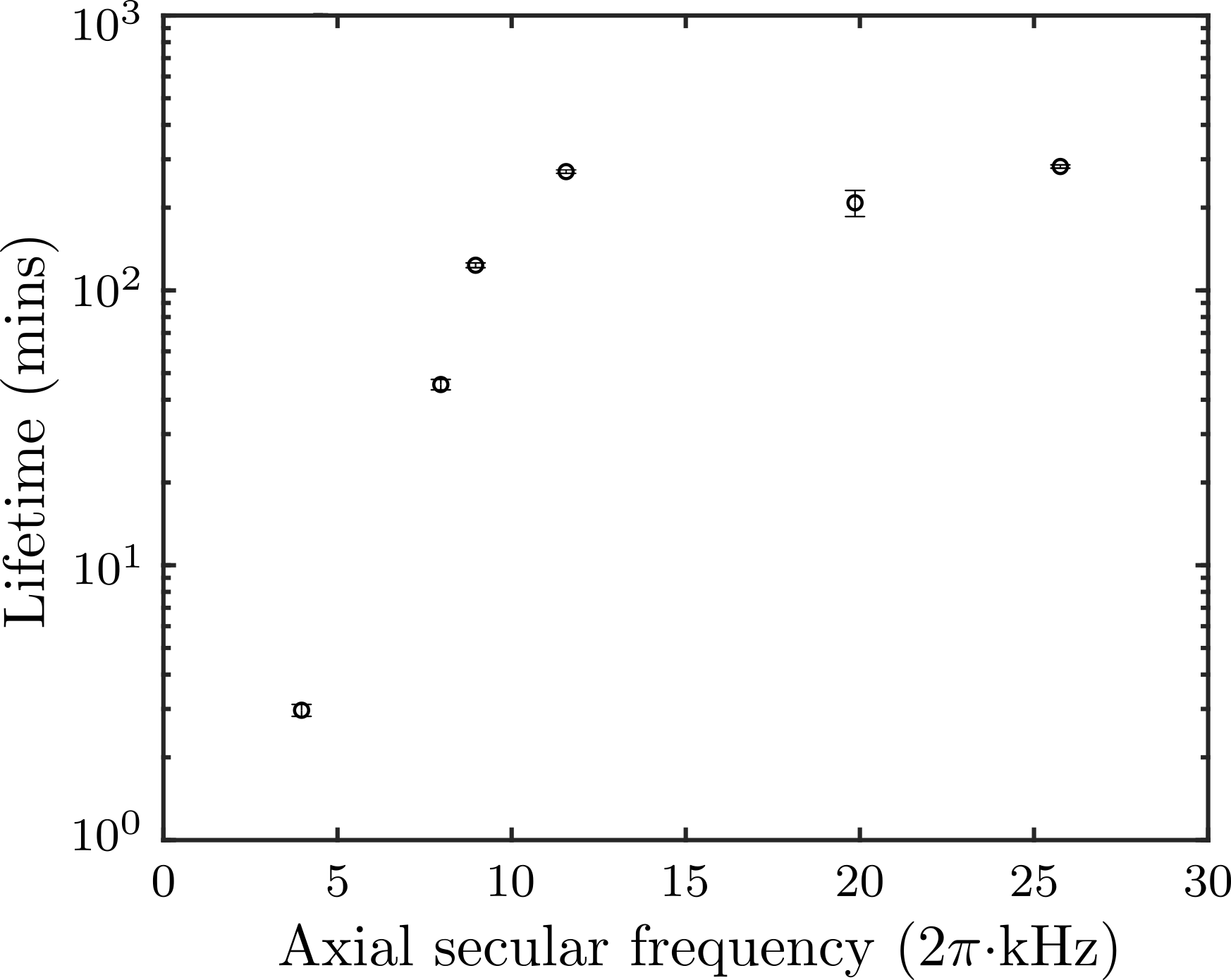
The lifetime of ions in the trap is shown in Fig. 1 for several values of the axial secular frequency of a single trapped 138Ba+. The axial frequency is measured by applying an oscillating voltage to one of the endcaps, which resonantly excites the ion motion Home et al. (2011). When the axial secular frequency is larger than kHz, the micromotion-interruption-induced loss appears to be mostly suppressed due to the increased trap depth. In the subsequent experiments, the axial secular frequency is fixed to kHz.
With the role of micromotion interruption clarified, the remaining trap loss is presumed to be predominantly due to chemical reactions with the residual background gas that can, potentially, both release a large amount of energy and convert the atomic ion into a molecular compound. To investigate this, we expose a trapped sample of 138Ba+ to one of three common UHV residual gases H2, CO2, and H2O, and monitor the trap loss – although CO is also a common residual gas it does not react with Ba+ Roth et al. (2008) and is not studied here. Using an integrated time-of-flight mass spectrometer (ToF-MS) Schowalter et al. (2012), we monitor the appearance of any ion products of the chemical reactions.
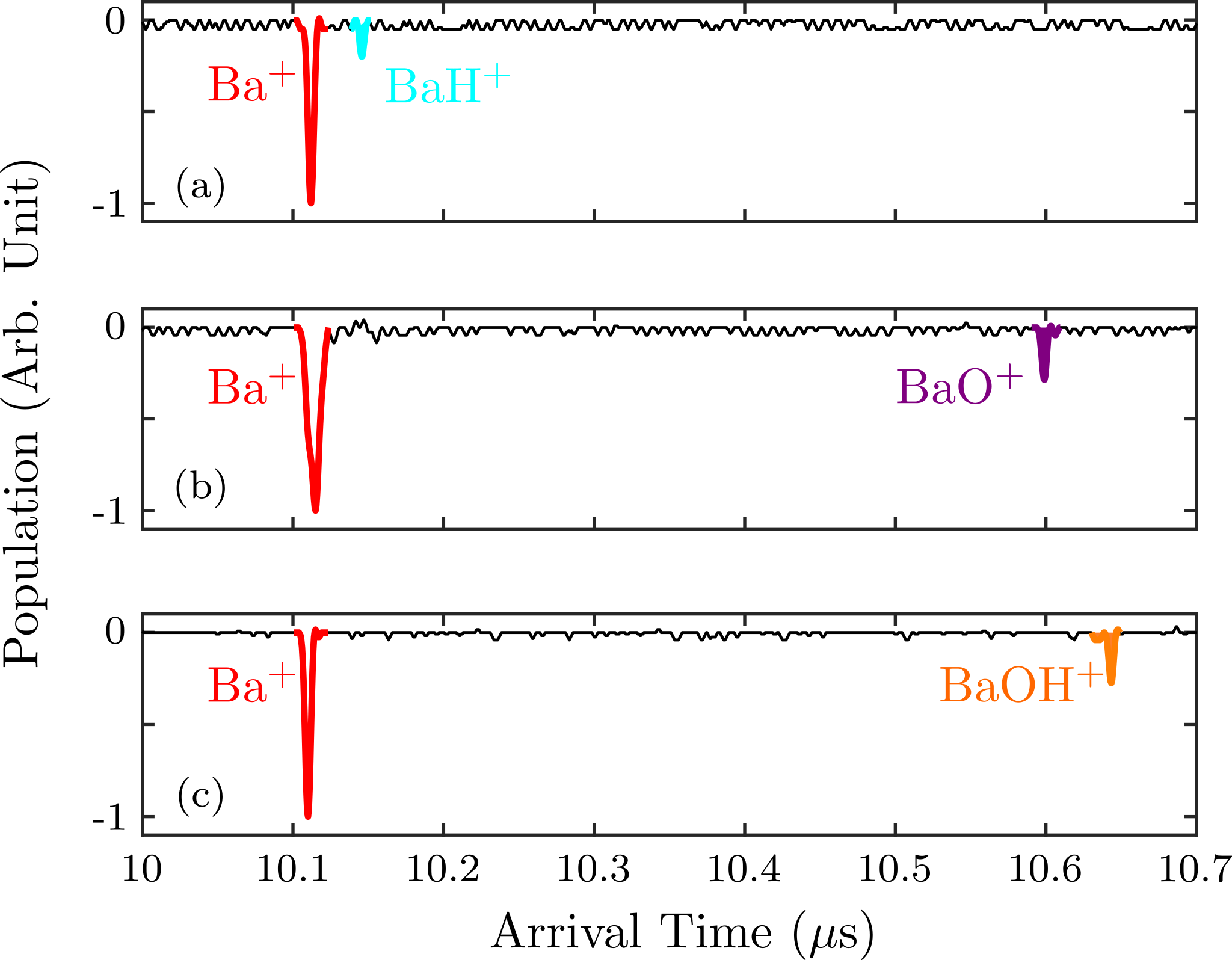
For the case of H2, a small 138Ba+ Coulomb crystal is prepared and H2 is introduced into the chamber with partial pressure of 10-8 mbar. Typically after approximately 30 mins, one of the ions in the crystal will become dark as shown in middle panel of Fig. 2. At this point, the ions are ejected into the ToF-MS revealing that, as shown in Fig.3(a), the dark ion is BaH+, presumably produced via the reaction
| (1) |
As this reaction is endoergic by eV and eV for 138Ba+ in 2S1/2 and 2D3/2 state, respectively, it likely proceeds via the 138Ba+ (2P1/2) electronic state accessed during laser cooling. In this case, the reaction releases eV, of which 99.3% is carried away by the light H atom, resulting in a BaH+ molecular ion that remains embedded in the Coulomb crystal. This production pathway may be of use to experiments aiming to leverage the large rovibrational constants of BaH+ to facilitate initialization into a single quantum state to search for the variation in mp/me Kajita et al. (2011).

The reaction with CO2 is studied in a similar manner. CO2 is introduced into the chamber at a partial pressure of mbar. One ion in the crystal will react and turn dark typically within 5 mins. The reaction rate constant with CO2 is around 100 higher than that of H2. Ejection into the ToF-MS reveals that the dark ions are BaO+, as shown in Fig. 3(b), presumably formed from the reaction
| (2) |
This reaction can proceed for both the electronic 2P1/2 and 2D3/2 states of 138Ba+, which release an energy of eV and eV, respectively Murad (1982); Roth et al. (2008). Due to the heavier mass of the CO product, the BaO+ can receive significantly more kinetic energy than the BaH+. The excess kinetic energy ( eV) must be dissipated via sympathetic cooling from the remaining laser-cooled 138Ba+ for the recoiling product ion to be recaptured in the crystal. Otherwise, the ion is likely to stay in a large orbit and experience heating due to the further collisions until it is eventually lost from the trap.
Finally, the reaction with H2O is studied in a similar apparatus used for molecular ion spectroscopy West . After baking, the H2O background pressure is suppressed below 10-10 mbar. A small amount of H2O can be released by ablating a barium chloride dihydrate (BaCl2(H2O)2) pellet. Under this condition, one or more BaOH+ are typically prepared in a 138Ba+ ion chain, revealed by the ToF-MS. These molecular ions are presumably formed from the reaction
| (3) |
This reaction can proceed for the electronic 2S1/2, 2P1/2 and 2D3/2 states of 138Ba+, which release an energy of eV, eV, and eV, respectively, of which 99.4% is carried away by the H product atom.
Anecdotally, we observe that while the BaH+/BaOH+ product is maintained in the trap at low axial secular frequency ( kHz), BaO+ is only contained for higher axial secular frequency and/or within a 3D Coulomb crystal, which provides better sympathetic cooling than a linear ion chain. In fact at the maximum axial secular frequency here ( kHz) we estimate the efficiency of recapturing the BaO+ product in a 12-ion crystal is %. While the recoiling BaO+ product does not have enough energy to escape the ion trap, it presumably resides in a large orbit that is relatively decoupled from the laser-cooled ion crystal. Here, further collisions or reactions can cause it to be lost from the trap. We occasionally observe the ion reappearing in the trap after several minutes. This suggests that with tighter trapping, and therefore improved sympathetic cooling from the remaining laser-cooled ions Guggemos et al. (2015), all of the molecular ion reaction products can be maintained in the trap, thus opening the possibility of using photodissociation to reclaim the parent ion and avoid reloading the ion trap.
To explore this possibility, it is necessary to determine potential photodissociation wavelengths for all three molecule systems. For BaH+, Ref. Mejrissi et al. (2013) suggests two useful paths for photodissociation. First, a laser with wavelength nm is able to drive BaH+ from low-lying rovibrational states of the ground X state to the unbound A state. Second, a laser with wavelength nm can excite BaH+ from the low-lying rovibrational states of the ground X state to the unbound C state Mejrissi et al. (2013). Both pathways result in dissociation to a 138Ba+ and H, potentially allowing the parent 138Ba+ ion to be recovered.
For recovery of Ba+ from BaO+ via photodissociation, we perform ab initio electronic structure calculations for the higher-energy excited states to predict transition energies to dissociative states. The details of these calculations are given in a previous publication Bartlett et al. (2015), and we note here only the difference for the current study. In this case, we use the augmented valence triple zeta basis set for the O atom Woon and Dunning (1993). The lowest energy repulsive state is found to be 2, which can be reached from the ground state using light in the 210-228 nm range with a transition moment of approximately 0.47 D.
For recovery of Ba+ from BaOH+, the dissociation energy from a combined theoretical and experimental determination was reported to be within the range from 225 to 243 nm Rossa et al. (2012).
Ideally, a single laser wavelength could be used to dissociate these products so that regardless of the background gas reactant the 138Ba+can be recovered. Taking the intersection of the available photodissociation wavelengths suggests that light around 225 nm should be sufficient for recovering Ba+ in all of the aforementioned cases. Therefore, we introduce a single pulsed-dye laser beam at 225 nm (10 ns pulse, 3 mW/mm2 on average) into the apparatus along the axial direction of the ion trap.
A typical sequence for recovery of Ba+ from, e.g., BaH+ using this laser is shown in Fig. 2. A 12-ion crystal is prepared and exposed to H2 for 30 mins before a BaH+ ion appears, Fig. 2(b). Next, the photodissociation laser is introduced and the BaH+ is dissociated and the parent Ba+ is recovered. We have also observed that a 370 nm continuous wave laser with a laser intensity of 2 mW/mm2 recovers the Ba+ from BaH+. The pathway for dissociation with this laser is presumably a transition from the low-lying rovibrational states of X to A states above the dissociation threshold Mejrissi et al. (2013).
For BaO+ and BaOH+ we observe similar behavior. Once dark ions appear, the photodissociation laser recovers the parent ion. In the case of BaO+, we observe that efficient photodissociation occurs for wavelengths ranging from 210 nm to 225 nm, suggesting that the technologically-convenient 5th harmonic of a Nd:YAG laser could be used in this case. For BaOH+ we observe efficient photodissociation for wavelengths ranging from 225 nm to 240 nm.
With a single laser capable of dissociating the products of reactions with typical UHV gases, it is possible to extend the effective trap lifetime by periodically recovering Ba+ from the produced molecular ions. To investigate this, the ion trap is illuminated with the photodissociation laser at 10 Hz repetition rate. Fig. 4 shows the lifetime enhancement of trapped 138Ba+ due to the presence of the photodissociation laser. Once the photodissociation laser is off, the UHV background gas limits the lifetime of a 12-ion chain of 138Ba+ to 290(13) mins, represented by the black circles in Fig. 4. The presence of the photodissociation laser extends the lifetime to 680(20) mins, as indicated by the red squares in Fig. 4.
The lifetime enhancement is presumably limited by the % probability of capturing the BaO+ products into the ion chain. To explore that, a 500-ion crystal of 138Ba+ is prepared, which can provide better sympathetic cooling and improve the trapping efficiency of recoiling product molecular ions. Without the UV dissociation laser present, the crystal represented by the dotted line in Fig. 4 shows a similar lifetime to that of the ion chain. The dashed line in Fig. 4 shows the lifetime of the crystal is extended to 2600(220) mins, further supporting the conclusion that the ion chain trap lifetime is limited by the efficiency of recapturing BaO+. As shown in Ref. Guggemos et al. (2015), if the dark ions are produced in the trap center, the sympathetic cooling time is inversely proportional to the cube of the secular frequency. Thus, traps with higher secular frequency should expect significantly higher trap-lifetime improvements with the 225 nm photodissociation laser present.

Like Ba+, other trapped ion species can also react with the common UHV background gases, e.g. H2, CO, CO2 and H2O. Table 1 shows a summary of possible reaction products and corresponding dissociation wavelengths for the products in their vibrational ground states.
While only Ba+ appears to have the fortuitous overlapping of all photodissociation pathways, the laser used for laser cooling of Ca+ or Yb+ dissociates one of the products“automatically” meaning that only one additional laser is needed to dissociate all products. Further, a properly baked UHV chamber will typically present a background gas primarily composed of H2, CO, and CO2, which would lead to only the hydride and oxide products. As shown in column 4 of Tab. 1, there are technologically convenient pathways for dissociating these products for Mg+, Ca+, Ba+, and Yb+.
Of course, it is preferable to prevent these reactions from ever occurring. While H2 outgassing is typically assumed to be primarily responsible for ion loss as it tends to be the predominate residual gas, the reaction rate constants of CO and CO2 may be considerably higher than that of H2 – e.g. here we find that the reaction with Ba+ ions is around 100 faster for CO2 than H2 – and all of these loss channels may play a significant role. As a result, it is important to suppress the outgassing rates of H2, CO, and CO2. In general, outgassing rates can be suppressed either by vacuum firing at K or by an air bake at K, both of which lead to the building of an non-permeable oxide layer on the interior of the vacuum vessel Bernardini et al. (1998). Ref. Mahner et al. (2003) demonstrates that the CO2 desorption from the stainless steel chamber can also be suppressed through sputter coating of nonevaporable getter film followed by K baking.
| Dissociation Wavelength | All products | oxides & hydrides | |
| (nm) | (nm) | (nm) | |
| BeH+ | 140-210 Yang et al. (2020)/157 Sawyer et al. (2015) | ||
| BeO+ | 273-315 Ghalila et al. (2008) | N/A | N/A |
| BeOH+ | 223-236a | ||
| MgH+ | 173-193/281 Højbjerre et al. (2009) | ||
| MgO+ | 156-176/368-478 Maatouk et al. (2011) | N/A | 173-176 |
| MgOH+ | 324-383a | ||
| CaH+ | 283-287 Kimura et al. (2017)/370-421 Rugango et al. (2016) | Cooling laser + | |
| CaO+ | 318-375 Khalil et al. (2013) | N/A | 318-375 |
| CaOH+ | 233-255 | ||
| SrH+ | 240-270 Abu el kher et al. (2021) | ||
| SrO+ | 208-226a | N/A | N/A |
| SrOH+ | 205-220a | ||
| BaH+ | 225-265/370-450 Mejrissi et al. (2013) | ||
| BaO+ | 210-228 Bartlett et al. (2015) | 225-228 | 225-228 |
| BaOH+ | 225-243 Rossa et al. (2012) | ||
| YbH+ | 369 Sugiyama and Yoda (1997)/405 Hoang et al. (2020) | Cooling laser + | Cooling laser + |
| YbO+ | 188-200/257-286a | 257-286a | 257-286a |
| YbOH+ | 216-330a |
-
•
a. the relevant dissociation wavelengths are estimated in this work.
In summary, we observed that the trap lifetime of Ba+ ions is limited by chemical reaction with some common UHV background gases (H2, CO, CO2 and H2O) once the micromotion-interruption -induced loss is suppressed with high-enough trap depth. Using relevant spectroscopic data and ab initio molecular structure calculations, we identified photodissociation pathways for all of the produced molecular ions and recovered the parent atomic ion from the product molecular ions with a single laser. Specifically, we observed an improvement in the effective trap lifetime by and for an ion chain and ion crystal, respectively, with a 225 nm laser. The data suggest that the lifetime enhancement is limited by the efficiency of BaO+ recapture, which should improve significantly with a larger secular frequency as employed in most trapped ion quantum logic experiments. This process should be straightforward to extend to other atomic ion species and the requisite dissociation wavelengths are provided. This technique could be employed in trapped ion quantum logic experiments to recover lost ions and avoid reloading of the ion trap.
Acknowledgements
We acknowledge support from the NSF QLCI program through grant number OMA-2016245. This work was also supported in part by National Science Foundation (Grants No. PHY-1255526, No. PHY-1415560, No. PHY-1912555, No. CHE-1900555, and No. DGE-1650604) and Army Research Office (Grants No. W911NF-15-1-0121, No. W911NF-14-1-0378, No. W911NF-13-1-0213 and W911NF-17-1-0071) grants.
References
- Bruzewicz et al. (2019a) C. D. Bruzewicz, J. Chiaverini, R. McConnell, and J. M. Sage, Applied Physics Reviews 6, 021314 (2019a).
- Christensen et al. (2020) J. E. Christensen, D. Hucul, W. C. Campbell, and E. R. Hudson, npj Quantum Information 6, 35 (2020).
- Ballance et al. (2016) C. J. Ballance, T. P. Harty, N. M. Linke, M. A. Sepiol, and D. M. Lucas, Phys. Rev. Lett. 117, 060504 (2016).
- (4) R. Srinivas et al., arXiv:2102.12533 .
- Gaebler et al. (2016) J. P. Gaebler, T. R. Tan, Y. Lin, Y. Wan, R. Bowler, A. C. Keith, S. Glancy, K. Coakley, E. Knill, D. Leibfried, and D. J. Wineland, Phys. Rev. Lett. 117, 060505 (2016).
- Bruzewicz et al. (2019b) C. D. Bruzewicz, R. McConnell, J. Stuart, J. M. Sage, and J. Chiaverini, npj Quantum Information 5, 102 (2019b).
- Mehta et al. (2020) K. K. Mehta, C. Zhang, M. Malinowski, T.-L. Nguyen, M. Stadler, and J. P. Home, Nature 586, 533 (2020).
- Bruzewicz et al. (2016) C. D. Bruzewicz, R. McConnell, J. Chiaverini, and J. M. Sage, Nature Communications 7, 13005 (2016).
- Pagano et al. (2018) G. Pagano, P. W. Hess, H. B. Kaplan, W. L. Tan, P. Richerme, P. Becker, A. Kyprianidis, J. Zhang, E. Birckelbaw, M. R. Hernandez, Y. Wu, and C. Monroe, Quantum Science and Technology 4, 014004 (2018).
- Brandl et al. (2016) M. F. Brandl, M. W. van Mourik, L. Postler, A. Nolf, K. Lakhmanskiy, R. R. Paiva, S. Möller, N. Daniilidis, H. Häffner, V. Kaushal, T. Ruster, C. Warschburger, H. Kaufmann, U. G. Poschinger, F. Schmidt-Kaler, P. Schindler, T. Monz, and R. Blatt, Review of Scientific Instruments 87, 113103 (2016).
- Leopold et al. (2019) T. Leopold, S. A. King, P. Micke, A. Bautista-Salvador, J. C. Heip, C. Ospelkaus, J. R. Crespo López-Urrutia, and P. O. Schmidt, Review of Scientific Instruments 90, 073201 (2019).
- Sawyer et al. (2015) B. C. Sawyer, J. G. Bohnet, J. W. Britton, and J. J. Bollinger, Phys. Rev. A 91, 011401 (2015).
- Chen et al. (2011) K. Chen, S. J. Schowalter, S. Kotochigova, A. Petrov, W. G. Rellergert, S. T. Sullivan, and E. R. Hudson, Phys. Rev. A 83, 030501 (2011).
- Guggemos et al. (2015) M. Guggemos, D. Heinrich, O. A. Herrera-Sancho, R. Blatt, and C. F. Roos, New Journal of Physics 17, 103001 (2015).
- Chen et al. (2014) K. Chen, S. T. Sullivan, and E. R. Hudson, Phys. Rev. Lett. 112, 143009 (2014).
- Chen et al. (2013) K. Chen, S. T. Sullivan, W. G. Rellergert, and E. R. Hudson, Phys. Rev. Lett. 110, 173003 (2013).
- Schowalter et al. (2016) S. J. Schowalter, A. J. Dunning, K. Chen, P. Puri, C. Schneider, and E. R. Hudson, Nature Communications 7, 12448 (2016).
- Cetina et al. (2012) M. Cetina, A. T. Grier, and V. Vuletić, Phys. Rev. Lett. 109, 253201 (2012).
- Mills (2020) M. Mills, Ph.D. thesis, University of California Los Angeles (2020).
- Home et al. (2011) J. P. Home, D. Hanneke, J. D. Jost, D. Leibfried, and D. J. Wineland, New Journal of Physics 13, 073026 (2011).
- Roth et al. (2008) B. Roth, D. Offenberg, C. B. Zhang, and S. Schiller, Phys. Rev. A 78, 042709 (2008).
- Schowalter et al. (2012) S. J. Schowalter, K. Chen, W. G. Rellergert, S. T. Sullivan, and E. R. Hudson, Review of Scientific Instruments 83, 043103 (2012).
- Kajita et al. (2011) M. Kajita, M. Abe, M. Hada, and Y. Moriwaki, Journal of Physics B: Atomic, Molecular and Optical Physics 44, 209802 (2011).
- Murad (1982) E. Murad, The Journal of Chemical Physics 77, 2057 (1982).
- (25) E. West, In preparation.
- Mejrissi et al. (2013) L. Mejrissi, H. Habli, H. Ghalla, B. Oujia, and F. X. Gadéa, The Journal of Physical Chemistry A 117, 5503 (2013), pMID: 23701525.
- Bartlett et al. (2015) J. H. Bartlett, R. A. VanGundy, and M. C. Heaven, The Journal of Chemical Physics 143, 044302 (2015).
- Woon and Dunning (1993) D. E. Woon and T. H. Dunning, The Journal of Chemical Physics 98, 1358 (1993).
- Rossa et al. (2012) M. Rossa, I. Cabanillas-Vidosa, G. A. Pino, and J. C. Ferrero, The Journal of Chemical Physics 136, 064303 (2012).
- Bernardini et al. (1998) M. Bernardini, S. Braccini, R. De Salvo, A. Di Virgilio, A. Gaddi, A. Gennai, G. Genuini, A. Giazotto, G. Losurdo, H. B. Pan, A. Pasqualetti, D. Passuello, P. Popolizio, F. Raffaelli, G. Torelli, Z. Zhang, C. Bradaschia, R. Del Fabbro, I. Ferrante, F. Fidecaro, P. La Penna, S. Mancini, R. Poggiani, P. Narducci, A. Solina, and R. Valentini, Journal of Vacuum Science & Technology A 16, 188 (1998).
- Mahner et al. (2003) E. Mahner, J. Hansen, J.-M. Laurent, and N. Madsen, Phys. Rev. ST Accel. Beams 6, 013201 (2003).
- Yang et al. (2020) Y. K. Yang, Y. Cheng, Y. G. Peng, Y. Wu, J. G. Wang, Y. Z. Qu, and S. B. Zhang, Journal of Quantitative Spectroscopy and Radiative Transfer 254, 107203 (2020).
- Ghalila et al. (2008) H. Ghalila, S. Lahmar, Z. B. Lakhdar, and M. Hochlaf, Journal of Physics B: Atomic, Molecular and Optical Physics 41, 205101 (2008).
- Højbjerre et al. (2009) K. Højbjerre, A. K. Hansen, P. S. Skyt, P. F. Staanum, and M. Drewsen, New Journal of Physics 11, 055026 (2009).
- Maatouk et al. (2011) A. Maatouk, A. B. Houria, O. Yazidi, N. Jaidane, and M. Hochlaf, Journal of Physics B: Atomic, Molecular and Optical Physics 44, 225101 (2011).
- Kimura et al. (2017) N. Kimura, M. Kajita, and K. Okada, Journal of Physics: Conference Series 875, 022042 (2017).
- Rugango et al. (2016) R. Rugango, A. T. Calvin, S. Janardan, G. Shu, and K. R. Brown, Chemphyschem : a European journal of chemical physics and physical chemistry 17, 3764—3768 (2016).
- Khalil et al. (2013) H. Khalil, F. Le Quéré, C. Léonard, and V. Brites, The Journal of Physical Chemistry A 117, 11254 (2013).
- Abu el kher et al. (2021) N. Abu el kher, I. Zeid, N. El-Kork, and M. Korek, Journal of Computational Science 51, 101264 (2021).
- Sugiyama and Yoda (1997) K. Sugiyama and J. Yoda, Phys. Rev. A 55, R10 (1997).
- Hoang et al. (2020) T. M. Hoang, Y.-Y. Jau, R. Overstreet, and P. D. D. Schwindt, Phys. Rev. A 101, 022705 (2020).