Free Energy Cost to Assemble Superlattices of Polymer-Grafted Nanoparticles
Abstract
Mesoparticles consisting of a hard core and a soft corona like polymer-grafted nanoparticles (PGNPs) can assemble into various superlattice structures, in which each mesoparticle assumes the shape of the corresponding Wigner-Seitz (or Voronoi) cell. Conventional wisdom often perceives the stability of these superlattices in a mean-field view of surface area minimization or corona entropy maximization, which lacks a molecular interpretation. We develop a simulation method to calculate the free energy cost to deform spherical PGNPs into Wigner-Seitz polyhedra, which are then relaxed in a certain crystalline superlattice. With this method, we successfully quantify the free energy differences between model BCC, FCC and A15 systems of PGNPs and identify BCC as the most stable structure in most cases. Analysis of polymer configurations in the corona, whose boundary is blurred by chain interpenetration, shows that the radial distribution of grafted chains and the corresponding entropy is almost identical among BCC and FCC, suggesting that the higher stability of BCC structure cannot be explained by a mean-field description of corona shape.
1 Introduction
Many complex properties of soft matter, such as ordered or disordered mesoscopic structures, weak interactions comparable to thermal agitation and strong response to weak perturbation, are rooted in the fact that the basic building blocks of the system are soft deformable mesoparticles Brochard-Wyart et al. (2020). Such soft mesoparticles, as diverse as polymer-grafted nanoparticles (PGNPs) Akcora et al. (2009), spherical micelles of diblock copolymers Thomas et al. (1987), giant supramolecules Ungar et al. (2003), ligand-capped nanocrystals Goodfellow et al. (2015), and DNA-functionalized nanoparticles Nykypanchuk et al. (2008), are all made of a relatively hard core and a soft corona of chain molecules. The chemical complexity in above examples provides us with plethora of possibilities to manipulate their self-assembly behavior and enables the fabrication of various superlattice structures including but not limited to body-centered cubic (BCC), face-centered cubic (FCC) and hexagonal-closed packed Si et al. (2018); Girard et al. (2019). A quantitative account of the stability of competing superlattices is of both theoretical and experimental interest, which is the prerequisite to predict equilibrium phase properties Boles et al. (2016).
Although packing has been successfully applied to describe hard particles Cersonsky et al. (2018), it does not capture the effects of soft corona. Attempts have been made to model soft mesoparticles with simple effective potentials such as hard-core square shoulder (HCSS) potential Ziherl and Kamien (2000, 2001); Pattabhiraman and Dijkstra (2017) and inverse power law potential Travesset (2015). Theoretical analysis based on simple mean-field picture identifies two contributions, lattice vibration and surface minimization, to the overall crystal stability Ziherl and Kamien (2000, 2001). However, interactions in these models are spherically symmetric and pair-wise additive, thus miss the intrinsic anisotropy and manybody effects of core-corona mesoparticles. In fact, one key feature of these deformable mesoparticles is that they adopt the shape of Wigner-Seitz cells (or Voronoi cells) in the superlattice Goodfellow et al. (2015); Bilchak et al. (2017); Reddy et al. (2018). These polyhedral unit cells of soft mesoparticles are analogously observed in metals Lifshitz (2014) and metallic nanocrystals Li et al. (2020), but due to different physical origins.
To address the polyhedral shapes of soft mesoparticles, another important perspective was introduced, which can date back to the famous Kelvin problem on dividing space into equal partitions with minimum amount of materials Williams (1968). Regarding the Wigner-Seitz polyhedron of BCC (truncated octahedron) and FCC (rhombic dodecahedron), the surface area is less than by for a given volume. Kelvin’s original solution was a modified truncated octahedron where some faces are changed to be curved. It was later recognized that, by allowing two types of polyhedra, the minimal area solution to the Kelvin’s problem becomes the Weaire-Phelan foam, which contains six tetrakaidecahedra (Z14) and two irregular dodecahedra (Z12) in each unit cell Weaire and Phelan (1994); Kusner and Sullivan (1996). This structure actually occurs in nature as in A15 clathrate Rivier (1994), which belongs to the large group of Frank-Kasper phases Frank and Kasper (1959); Montis et al. (2021). The average surface area of the eight polyhedra in A15 unit cell is less than by Kusner and Sullivan (1996).
The minimal surface consideration has found its applicaiton in diblock copolymers, which organize into soft micelles to form various superlattice structures Grason et al. (2003). This soft micelle differs from other core-corona mesoparticles by its soft core made of one type of polymer segements. At different conditions, the shape of the soft core may change from spherical to polyhedral Reddy et al. (2018). The stability of diblock copolymer phases is then a tradeoff between the interfacial energy (between two blocks) and the stretching energy of chains Grason et al. (2003). Although BCC spherical phase was first discovered in diblock copolymers, unexpected Frank-Kasper phases are also observed Lee et al. (2010, 2014); Kim et al. (2017); Schulze et al. (2017); Watanabe et al. (2020). Self-consistent field theory (SCFT) predicts that conformation asymmetry and copolymer architecture (branched or not) can be tuned to favor the formation of complicated Frank-Kasper phases Liu et al. (2016); Li et al. (2017); Bates et al. (2019). However, a molecular-level understanding of the competition between different superlattice structures is still missing.
To resolve the stability of superlattices formed by deformable mesoparticles, we propose a free energy calculation scheme based on thermodynamic integration Frenkel and Smit (2002) and apply it on polymer-grafted nanoparticles (PGNPs). Similar free energy calculations have been performed on hydrocarbon-capped nanocrystals using thermodynamic perturbation Kaushik and Clancy (2013) and integration over pressure Zha and Travesset (2018). In our method, the free energy cost to assemble isolated spherical PGNPs into BCC, FCC and A15 superlattices are calculated in two steps. In step I, PGNPs are compressed into polyhedral shapes by confining walls. In step II, compressed PGNPs are relaxed on a superlattice while confining walls are removed. By implementing this methodology with coarse-grained molecular dynamics (MD) simulation, we quantify the stability of different superlattices in the limit of low (super)lattice vibration. We find that BCC structure is marginally more stable than FCC by for most volume studied. A test on one A15 structure shows it is more stable than FCC but still less stable than BCC, suggesting that minimal surface area does not always guarantee lower free energy. Close examination of polymer distribution around the nanoparticle core reveals that chain interpenetration from two PGNPs blurs the boundary of Wigner-Seitz polyhedra Midya et al. (2020). The corresponding entropy of corona chain distribution is almost indistinguishable among FCC and BCC Goodfellow et al. (2015), making the determination of superlattice stability by alone impossible.
2 Methods
2.1 Thermodynamic Integration
Thermodynamic integration is a standard free energy calculation method which connects the reference state “0” with the state of interest “1” by a reversible path denoted by a parameter, e.g. Frenkel and Smit (2002). If the potential energy at the two endpoints are and , then the potential energy for the coupled system at intermediate can be defined as and the free energy of interest is calculated by the integral along the path
| (1) |
where means to take ensemble average in the system interacting with potential energy .
We choose isolated PGNPs as our reference state “0” and consider a reversible thermodynamic path to assemble crystalline superlattices of PGNPs in two steps (Fig. 1). In step I, each spherical PGNP in isolated state is slowly compressed into a polyhedron (usually with a shape of the Wigner-Seitz cell) by gradually increasing the repulsion strength of confining walls. In step II, confined PGNPs are stacked to form the desired superlattice, whose interactions are slowly turned on while wall confinement is being removed. In this work, we do not calculate the reference free energy for the hyperthetical crystal formed by uncompressed spherical PGNPs. We also neglect the contribution from collective vibrations of PGNPs on the superlattice, which should be small for highly compressed states. Therefore, the free energy reported in this work is the cost (per PGNP) to assemble the superlattice from uncompressed spherical PGNPs, neglecting lattice vibration. We break into two terms corresponding to the two steps (to be explained below)
| (2) |
![[Uncaptioned image]](https://cdn.awesomepapers.org/papers/dbb47a18-55c6-4f5e-8baa-162abef57ba1/x1.png)
In step I, the starting point is the system of one isolated spherical PGNP with volume and potential energy , where is the energy of covalent bonds in polymer chains and is pairwise-additive non-bonded interactions between monomers or nanoparticles. During compression along the path, the PGNP experices repulsion from several confining walls adopting the corresponding geometry of the desired superlattice. The positions of these walls define the final volume of the PGNP after compression. The coupled potential energy at integration parameter is defined as
| (3) |
with changing from to . The free energy cost in this step can be roughly associated with volume compression of the PGNP and is calculated by the integral
| (4) |
In step II, the fully compressed PGNPs from step I are packed into the desired superlattice. Non-bonded interactions in the system can be decomposed into three parts, , where is the interaction energy among the particles of the central PGNP, is the interaction energy among the particles of the surrounding PGNPs (usually just first nearest neighbors), and is interaction energy between particles of the central PGNP and particles of the surrounding PGNPs. In addition to the walls confining the central PGNP (same as in step I), an equal number of extra walls facing outward are needed to confine the surrounding PGNPs. There is no wall between two surrounding PGNPs. If the interaction with all the walls is , then the coupled potential energy is written as
| (5) |
with changing from to . In practice, one might need to start from over-compressed PGNPs with smaller volume , which leave small gaps between them to avoid initial overlap of polymer chains. After relaxation, these gaps are filled by polymer chains. The contribution of the free energy in step II to the total free energy cost per PGNP is
| (6) |
which is related to the relaxation of surfaces between PGNPs after walls are removed. The factor is due to the sharing of each surface by two PGNPs.
2.2 Models and Molecular Dynamics Simulation
We use the coarse-grained model of polymer chains and nanoparticles, in which non-bonded interactions between a pair of monomers () and/or nanoparticles () at a distance apart follow the repulsive shifted Lennard-Jones (LJ) potential, when , where is the unit of energy and the monomer size is the unit of length Bilchak et al. (2017). The choice of , and makes the effective size of bare nanoparticles roughly . The cutoff distance is set to so that all pair interactions are purely repulsive. Polymers are first modeled as bead-spring chains connected by finite extensible nonlinear elastic (FENE) bonds, , with force constant and bond length Kremer and Grest (1990). We also consider semiflexible chains with the harmonic bond angle potential added on top of FENE bonds, where rad-2 and . We graft polymers by anchoring the first monomer of each chain on the surface of the nanoparticle with a rigid bond. The corresponding surface graft density is . Several chain lengths are studied with the number of monomers per chain .
![[Uncaptioned image]](https://cdn.awesomepapers.org/papers/dbb47a18-55c6-4f5e-8baa-162abef57ba1/box.png)
When PGNPs need to be confined into the desired polyhedral shape, polymers also interact with several repulsive walls around the PGNP through a harmonic potential , where is the penetration depth of a monomer into the wall surface. In a system with walls and monomers, the total wall energy is . For the BCC, FCC and A15 structures under study, walls are placed at the faces of the corresponding Wigner-Seitz cells, which are truncated octahedron (BCC), rhombic dodecahedron (FCC), tetrakaidecahedron and irregular dodecahedron (A15), respectively. Details of wall positions are described in Supporting Information. In step I, only one PGNP is needed under open boundary conditions. In step II, several surrounding PGNPs (not only nearest neighbors, but also some next nearest neighbors) are needed to enclose the central one. By properly choosing the simulation box under periodic boundary condisions, we use 16, 16 and 64 PGNPs to study BCC, FCC and A15, respectively (Fig 2).
The LAMMPS package is used to run constant volume molecular dynamics simulations at fixed temperature with a time step , where the mass is set the same for all particles Plimpton (1995). Each system is equilibrated for time steps before ensemble average is taken. To evaluate each thermodynamic integral numerically, - points are used within Gauss-Lobatto quadrature Press et al. (1992). Two independent runs are performed for each system, whose numerical discrepancy is an order of magnitude smaller than the free energy difference between different phases studied here. All free energy results are reported in unit of .
3 Results and discussion
3.1 Stability of crystalline superlattices
We calculate the free energy cost to assemble BCC, FCC and A15 superlattices at various Wigner-Seitz (Voronoi) cell volumes for PGNPs with flexible (FENE bonds) or semiflexible (FENE bonds plus bond angles) grafts. The volume of the polyhedron in superlattice is compared with the original volume of an isolated spherical PGNP, whose radius is estimated as the average of the location of the last monomer on each chain. The typical thermodynamic integration curves in step I and II are shown in Fig. 3. The curve drops rapidly, by orders of magnitude, close to the state without confining walls ( in step I and in step II). To capture this fast change, we use points in each integration. The calculated volume and surface free energy cost is reported as per PGNP value.
![[Uncaptioned image]](https://cdn.awesomepapers.org/papers/dbb47a18-55c6-4f5e-8baa-162abef57ba1/TIa.png)
![[Uncaptioned image]](https://cdn.awesomepapers.org/papers/dbb47a18-55c6-4f5e-8baa-162abef57ba1/TIb.png)


As expected, the volume cost increases as decreases because more work is needed to compress a spherical PGNP into a smaller polyhedron (Fig. 4a). The surface term is negative because it corresponds to the free energy gain (entropy increase) after the confined flat interface is relaxed, allowing interpenetration of polymers from two neighboring PGNPs. The overall free energy cost still increases monotonically with decreasing .
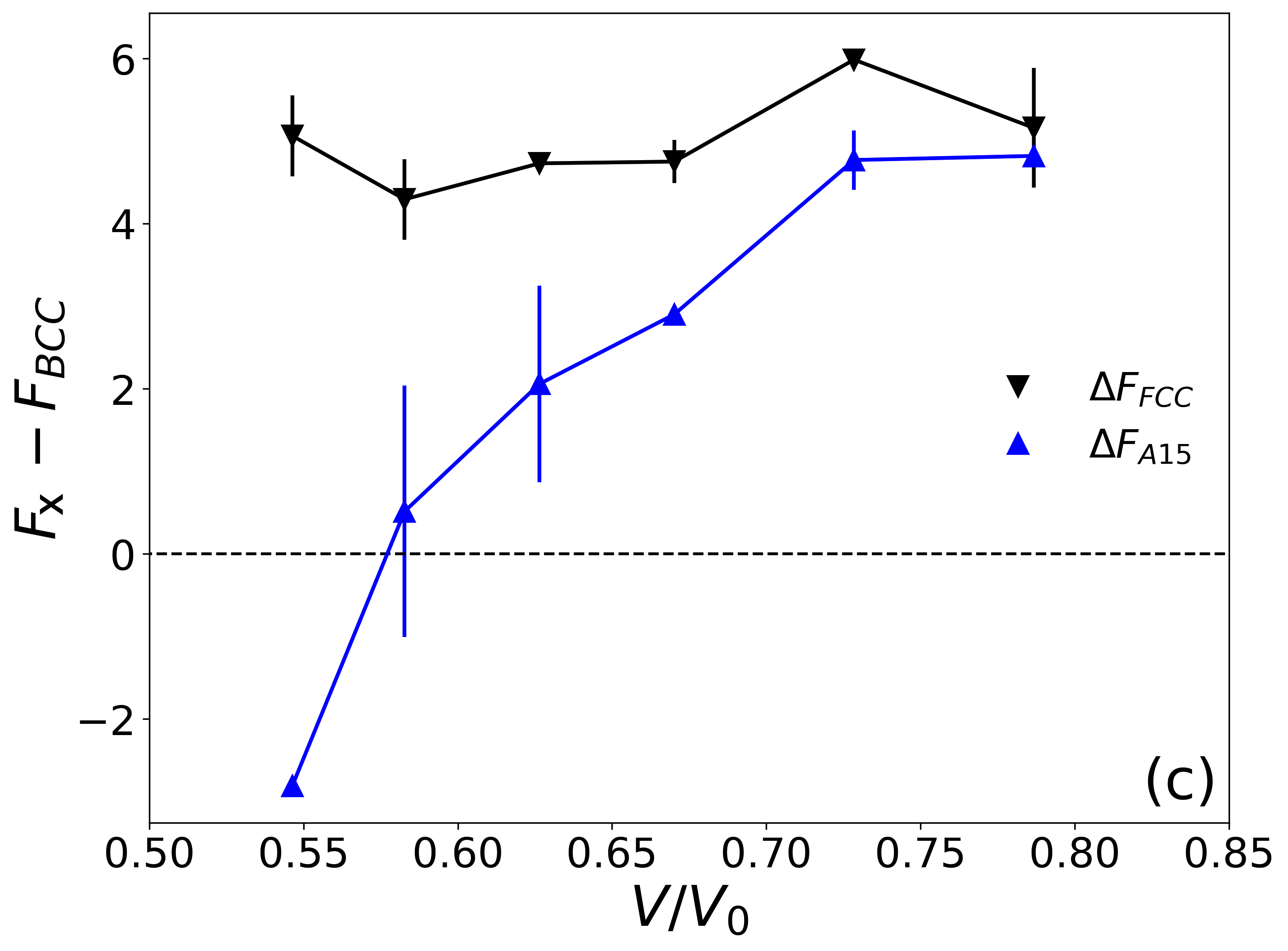
For all the chain lengths studied, being flexible or semiflexible, we find that BCC is more stable than FCC structure at large (Fig. 4b). We can first identify a higher free energy cost for confined FCC as shown in Fig. 4b, but the difference is only of . To compare the total free energy cost , we also need to include the free energy gain from surface relaxation. Since rhombic dodecahedron has more surface area than truncated octahedron, is slightly larger in FCC than BCC. But this surface relaxation is not strong enough to compensate the volume cost . Combining two effects, the free energy cost of BCC is lower than that of FCC structure by at most (see data in Supporting Information). This is in agreement with previous calculations on ligand-capped nanocrystals Zha and Travesset (2018), but in contrast to the theoretical analysis of entropy penalty for chain compression/extension, which estimates that BCC can be more stable than FCC in ligand-capped nanocrystals Goodfellow et al. (2015). In the small limit, the numerical results of of FCC is close to or even smaller than that of BCC. But the relative difference, , is on the order of , which is near the numerical precison of our thermodynamic integration scheme. Therefore, there is no clear evidence to claim that FCC is more stable than BCC at small volume, although it is possible.
We also examine the stability of A15 systems with short () flexible chains. Because the unit cell of A15 contains eight particles and 64 PGNPs are used in step II of thermodynamic integration, it is computationally too expensive to study longer chains under current protocol. In Voronoi tessellation of A15 structures, the two types of Frank-Kasper polyhedra, tetrakaidecahedron (Z14) and irregular dodecahedron (Z12), can have different volumes in principle. Previous study on block copolymers found that the free energy of A15 is optimized, when the volumes of Z14 and Z12 are slightly different Reddy et al. (2018). Here we focus on the equal-cell partition as in Weaire-Phelan foam Weaire and Phelan (1994), where the same for the Z14 and Z12 is assumed. The calculated the free energy shows that, although being slightly more stable than FCC, A15 is still less stable than BCC structure except at small (Fig. 4c). Since the surface area of the Wigner-Seitz polyhedron for a given volume obeys ( should be the weighted average of and ), this results suggests that the stability of different supperlattices does not simply follow the minimum-area rule as in foams Ziherl and Kamien (2001).
3.2 Distribution of monomers around nanoparticles
To reveal the underlying mechanisms for superlattice stability, we analyze the spatial distribution of polymer chains around nanoparticle cores. We first calculate monomer density profile along a certain crystallographic direction perpendicular to polyhedral faces. In either BCC or FCC, the inter-particle space is filled with monomers from the two neighboring PGNPs structured in three layers (Fig. 5). Close to the core, monomer density is higher than the bulk density and decays from the maximum value corresponding to the high surface grafting density . At intermediate distances, there is a “dry layer” with and monomers come from only one PGNP. Further away, coronas from two PGNPs start to overlap, forming an “interpenetration layer” Midya et al. (2020).
By symmetry, each shared polyhedral face bisects the center-to-center line at the location where monomer density from one PGNP equals to half of the total monomer density. This location can also be read from half of the lattice constant along each crystallographic direction (see Supporting Information for the value of ). Due to the interpenetration of chains, the faces of PGNP polyhedra on supperlattice are strongly roughened, and the shape of PGNPs is more smoothened and spherical than a perfect Wigner-Seitz polyhedron, as will be revealed below.

3.3 Distribution of end-to-end vector of grafted chains
We can quantify the states of grafted polymer chains by studying the distribution of their end-to-end vector , which points from the first anchoring monomer to the last monomer on each chain. The radial distribution for isolated spherical PGNPs, confined PGNPs under wall compression (after step I of thermodynamic integration) and relaxed PGNPs of our interest are shown in Fig. 6a. Here is the probability for the chain end to fall in the spherical shell beween radius and , regardless of chain orientation. For confined polyhedra (see inset in Fig. 3a), are more narrowly distributed. FCC has a slightly higher at and , but BCC has more chains with . This trend qualitatively agrees with the solid angle distribution for perfect truncated octahedron and rhombic dodecahedron Ungar et al. (2003); Goodfellow et al. (2015). In the study of ligand-capped nanocrystals Goodfellow et al. (2015), FCC Wigner-Seitz cell is considered to have a more anisotropic chain length (corona thickness) distribution, and to carry more entropy penalty than BCC when deformed from a hyperthetical sphere of the same volume . Although this argument about the entropy of chain length distribution may explain the higher stability of our confined BCC structure with a relatively regular polyhedron cell, it cannot resolve the stability of different relaxed structures as shown below.


We observe that, after wall confinement is removed, the distributions of relaxed BCC and FCC are almost indistinguishable (empty symbols in Fig. 6a), which agrees with our expectation that interpenetration of chains blurs the boundary of Wigner-Seitz polyhedra. Because the radial distribution of chain length alone does not even distinguish the shape of the two polyhedra, we need to study the orientational distribution of , i.e. of polar angle and of azimuthal angle . In Fig. 6b, we plot and for relaxed polyhedra of BCC and FCC. Surprisingly, both distributions have a larger deviation from the spherical case (black dotted lines) in BCC than in FCC. The distribution suggests that chains are more concentrated around the six square faces in truncated octahedron (), which separate the central PGNP from its second nearest neighbors. In these directions of BCC, the lattice constant is times of the nearest neighbor distance. In the case of rhombic dodecahedron, chains are slightly more clustered around , which correspond to the corner formed by four rhombus faces at a distance times of the nearest neighbor distance.
3.4 Entropy of end-to-end vector distribution
After obtaining the radial and angular , distribution of the end-to-end vector of grafted chains, we can compute the (Gibbs) entropy of the overall distribution for a given (relaxed) Wigner-Seitz polyhedron. If the total distribution of vector can be factored as , the corresponding normalization condition should be
By comparing with our three normalized distributions, , and , we can relate , and such that .
The entropy of the distribution can thus be expanded as
| (7) |
where
| (8) |
For spherically symmetric distributions, and (black dotted lines in Fig. 6b), which gives constant entropy terms and .
We fit the sampled distributions of relaxed BCC and FCC polyhedra with following functions (the trivial phase shift of is applied to the angle of FCC before the fitting)
| (9) |
The fitting curves for relaxed BCC and FCC are shown in Fig. 6, from which the entropy of the distribution is calculated as in Table 1.
| 18000 | 34000 | |||
|---|---|---|---|---|
| BCC | FCC | BCC | FCC | |
| 7.319 | 7.346 | 7.801 | 7.806 | |
| 1.833 | 1.837 | 1.830 | 1.837 | |
| 0.684 | 0.689 | 0.681 | 0.690 | |
| 9.833 | 9.879 | 10.300 | 10.319 | |
It can be seen that the total entropy and its radial or angular contributions of BCC structure are all lower or close to those of FCC. This disagrees with the previous knowledge that the higher stability of BCC superlattice can be explained by the entropy of chain length alone Goodfellow et al. (2015). It is worth emphasizing that chain interpenetration makes the radial distribution of chain length almost identical for BCC and FCC polyhedra. We thus argue that the free energy and stability of various superlattices should be dominated by energy and other entropy contributions, for instance, the configuration entropy of individual monomers.
3.5 Potential of mean force between two PGNPs
Although designed to calculate the free energy of superlattices of PGNPs, the current method can be straightforwardly modified to compute the potential of mean force (PMF) between a pair of PGNPs. Only one confining wall per PGNP is needed at the bisecting plane of the center-to-center line and only the two interacting PGNPs need to be considered under open boundary conditions in step II of the thermodynamic integration. Previously, the PMF has been estimated from the radial distribution function Striolo and Egorov (2007), interaction energy Verso et al. (2011) and force Meng et al. (2012) between two PGNPs. The similar problem has been studied in the context of polymer brush Milner et al. (1988); Milner (1991) and steric stabilization of colloidal particles Centeno et al. (2014).

We test this idea on a system with chain length by calculating the PMF as a function of the center-to-center distance . Obviously, the two PGNPs only start to experience a repulsion when , where is the radius of the isolated spherical PGNP with that chain length. Instead of the combined power law and inverse power law in terms of surface-to-surface distance (or brush height ) Milner et al. (1988); Centeno et al. (2014), our results show that the PMF can follow a simpler Hertzian-like repulsion with a fitting exponent Johnson (1987).
4 Conclusion
In this work, we present a simulation method that computes the free energy cost to deform a spherical PGNP into a Wigner-Seitz polyhedron followed by relaxation in a superlattice with certain crystalline symmetry. By applying this thermodynamic integration scheme on various systems, we confirm that BCC is more stable that FCC and A15 under most simulation conditions, but only by a small free energy difference. Comparison of polyhedral surface area and entropy of corona chain distribution suggests that none of these factors can explain the higher stability of BCC superlattice alone. The total free energy must be determined by the intricate interplay between energy and entropy under the superlattice geometry.
The current method can further be adapted to study the self-assembly of PGNPs in polymer melts Koh et al. (2020) or other deformable mesoparticles such as block copolymer micelles. As in the last example of PMF calculation, it would also be interesting to study polymer brush interactions or free energy cost to insert a nanoparticle into polymer brush Milchev et al. (2008) with our method.
References
- Brochard-Wyart et al. (2020) Françoise Brochard-Wyart, Pierre Nassoy, and Pierre-Henri Puech, Essentials of Soft Matter Science (CRC Press, 2020).
- Akcora et al. (2009) Pinar Akcora, Hongjun Liu, Sanat K Kumar, Joseph Moll, Yu Li, Brian C Benicewicz, Linda S Schadler, Devrim Acehan, Athanassios Z Panagiotopoulos, Victor Pryamitsyn, et al., “Anisotropic self-assembly of spherical polymer-grafted nanoparticles,” Nature Materials 8, 354–359 (2009).
- Thomas et al. (1987) Edwin L Thomas, David J Kinning, David B Alward, and Chris S Henkee, “Ordered packing arrangements of spherical micelles of diblock copolymers in two and three dimensions,” Macromolecules 20, 2934–2939 (1987).
- Ungar et al. (2003) Goran Ungar, Yongsong Liu, Xiangbing Zeng, Virgil Percec, and Wook-Dong Cho, “Giant supramolecular liquid crystal lattice,” Science 299, 1208–1211 (2003).
- Goodfellow et al. (2015) Brian W Goodfellow, Yixuan Yu, Christian A Bosoy, Detlef-M Smilgies, and Brian A Korgel, “The role of ligand packing frustration in body-centered cubic (bcc) superlattices of colloidal nanocrystals,” The Journal of Physical Chemistry Letters 6, 2406–2412 (2015).
- Nykypanchuk et al. (2008) Dmytro Nykypanchuk, Mathew M Maye, Daniel Van Der Lelie, and Oleg Gang, “Dna-guided crystallization of colloidal nanoparticles,” Nature 451, 549–552 (2008).
- Si et al. (2018) Kae Jye Si, Yi Chen, Qianqian Shi, and Wenlong Cheng, “Nanoparticle superlattices: the roles of soft ligands,” Advanced Science 5, 1700179 (2018).
- Girard et al. (2019) Martin Girard, Shunzhi Wang, Jingshan S Du, Anindita Das, Ziyin Huang, Vinayak P Dravid, Byeongdu Lee, Chad A Mirkin, and Monica Olvera de la Cruz, “Particle analogs of electrons in colloidal crystals,” Science 364, 1174–1178 (2019).
- Boles et al. (2016) Michael A Boles, Michael Engel, and Dmitri V Talapin, “Self-assembly of colloidal nanocrystals: From intricate structures to functional materials,” Chemical Reviews 116, 11220–11289 (2016).
- Cersonsky et al. (2018) Rose K Cersonsky, Greg van Anders, Paul M Dodd, and Sharon C Glotzer, “Relevance of packing to colloidal self-assembly,” Proceedings of the National Academy of Sciences 115, 1439–1444 (2018).
- Ziherl and Kamien (2000) Primož Ziherl and Randall D Kamien, “Soap froths and crystal structures,” Physical Review Letters 85, 3528 (2000).
- Ziherl and Kamien (2001) Primož Ziherl and Randall D Kamien, “Maximizing entropy by minimizing area: Towards a new principle of self-organization,” (2001).
- Pattabhiraman and Dijkstra (2017) Harini Pattabhiraman and Marjolein Dijkstra, “On the formation of stripe, sigma, and honeycomb phases in a core–corona system,” Soft Matter 13, 4418–4432 (2017).
- Travesset (2015) Alex Travesset, “Binary nanoparticle superlattices of soft-particle systems,” Proceedings of the National Academy of Sciences 112, 9563–9567 (2015).
- Bilchak et al. (2017) Connor R Bilchak, Eileen Buenning, Makoto Asai, Kai Zhang, Christopher J Durning, Sanat K Kumar, Yucheng Huang, Brian C Benicewicz, David W Gidley, Shiwang Cheng, et al., “Polymer-grafted nanoparticle membranes with controllable free volume,” Macromolecules 50, 7111–7120 (2017).
- Reddy et al. (2018) Abhiram Reddy, Michael B Buckley, Akash Arora, Frank S Bates, Kevin D Dorfman, and Gregory M Grason, “Stable frank–kasper phases of self-assembled, soft matter spheres,” Proceedings of the National Academy of Sciences 115, 10233–10238 (2018).
- Lifshitz (2014) Ron Lifshitz, “Explaining complex metals with polymers,” Proceedings of the National Academy of Sciences 111, 17698–17699 (2014).
- Li et al. (2020) XY Li, ZH Jin, X Zhou, and K Lu, “Constrained minimal-interface structures in polycrystalline copper with extremely fine grains,” Science 370, 831–836 (2020).
- Williams (1968) Robert Edward Williams, “Space-filling polyhedron: its relation to aggregates of soap bubbles, plant cells, and metal crystallites,” Science 161, 276–277 (1968).
- Weaire and Phelan (1994) Denis Weaire and Robert Phelan, “A counter-example to kelvin’s conjecture on minimal surfaces,” Philosophical Magazine Letters 69, 107–110 (1994).
- Kusner and Sullivan (1996) Rob Kusner and John M Sullivan, “Comparing the weaire-phelan equal-volume foam to kelvin’s foam,” Forma 11, 233–242 (1996).
- Rivier (1994) N Rivier, “Kelvin’s conjecture on minimal froths and the counter-example of weaire and phelan,” Philosophical Magazine Letters 69, 297–303 (1994).
- Frank and Kasper (1959) FC Frank and JS Kasper, “Complex alloy structures regarded as sphere packings. ii. analysis and classification of representative structures,” Acta Crystallographica 12, 483–499 (1959).
- Montis et al. (2021) Riccardo Montis, Luca Fusaro, Andrea Falqui, Michael B Hursthouse, Nikolay Tumanov, Simon J Coles, Terry L Threlfall, Peter N Horton, Rachid Sougrat, Anaïs Lafontaine, et al., “Complex structures arising from the self-assembly of a simple organic salt,” Nature 590, 275–278 (2021).
- Grason et al. (2003) Gregory M Grason, BA DiDonna, and Randall D Kamien, “Geometric theory of diblock copolymer phases,” Physical Review Letters 91, 058304 (2003).
- Lee et al. (2010) Sangwoo Lee, Michael J Bluemle, and Frank S Bates, “Discovery of a frank-kasper phase in sphere-forming block copolymer melts,” Science 330, 349–353 (2010).
- Lee et al. (2014) Sangwoo Lee, Chris Leighton, and Frank S Bates, “Sphericity and symmetry breaking in the formation of frank–kasper phases from one component materials,” Proceedings of the National Academy of Sciences 111, 17723–17731 (2014).
- Kim et al. (2017) Kyungtae Kim, Morgan W Schulze, Akash Arora, Ronald M Lewis, Marc A Hillmyer, Kevin D Dorfman, and Frank S Bates, “Thermal processing of diblock copolymer melts mimics metallurgy,” Science 356, 520–523 (2017).
- Schulze et al. (2017) Morgan W Schulze, Ronald M Lewis III, James H Lettow, Robert J Hickey, Timothy M Gillard, Marc A Hillmyer, and Frank S Bates, “Conformational asymmetry and quasicrystal approximants in linear diblock copolymers,” Physical Review Letters 118, 207801 (2017).
- Watanabe et al. (2020) Momoka Watanabe, Yusuke Asai, Jiro Suzuki, Atsushi Takano, and Yushu Matsushita, “Frank-kasper a15 phase formed in ab n block-graft copolymers with large numbers of graft chains,” Macromolecules 53, 10217–10224 (2020).
- Liu et al. (2016) Meijiao Liu, Yicheng Qiang, Weihua Li, Feng Qiu, and An-Chang Shi, “Stabilizing the frank-kasper phases via binary blends of ab diblock copolymers,” ACS Macro Letters 5, 1167–1171 (2016).
- Li et al. (2017) Weihua Li, Chao Duan, and An-Chang Shi, “Nonclassical spherical packing phases self-assembled from ab-type block copolymers,” (2017).
- Bates et al. (2019) Morgan W Bates, Joshua Lequieu, Stephanie M Barbon, Ronald M Lewis, Kris T Delaney, Athina Anastasaki, Craig J Hawker, Glenn H Fredrickson, and Christopher M Bates, “Stability of the a15 phase in diblock copolymer melts,” Proceedings of the National Academy of Sciences 116, 13194–13199 (2019).
- Frenkel and Smit (2002) D. Frenkel and B. Smit, Understanding Molecular Simulation (Academic Press, New York, 2002).
- Kaushik and Clancy (2013) Ananth P Kaushik and Paulette Clancy, “Solvent-driven symmetry of self-assembled nanocrystal superlattices–a computational study,” Journal of Computational Chemistry 34, 523–532 (2013).
- Zha and Travesset (2018) Xun Zha and Alex Travesset, “Stability and free energy of nanocrystal chains and superlattices,” The Journal of Physical Chemistry C 122, 23153–23164 (2018).
- Midya et al. (2020) Jiarul Midya, Michael Rubinstein, Sanat K Kumar, and Arash Nikoubashman, “Structure of polymer-grafted nanoparticle melts,” ACS Nano 14, 15505–15516 (2020).
- Kremer and Grest (1990) Kurt Kremer and Gary S Grest, “Dynamics of entangled linear polymer melts: A molecular-dynamics simulation,” The Journal of Chemical Physics 92, 5057–5086 (1990).
- Plimpton (1995) Steve Plimpton, “Fast parallel algorithms for short-range molecular dynamics,” Journal of Computational Physics 117, 1–19 (1995).
- Press et al. (1992) William H Press, William T Vetterling, Saul A Teukolsky, and Brian P Flannery, Numerical Recipes in C (Cambridge University Press, Cambridge, 1992).
- Striolo and Egorov (2007) Alberto Striolo and SA Egorov, “Steric stabilization of spherical colloidal particles: Implicit and explicit solvent,” The Journal of Chemical Physics 126, 014902 (2007).
- Verso et al. (2011) Federica Lo Verso, Leonid Yelash, Sergei A Egorov, and Kurt Binder, “Interactions between polymer brush-coated spherical nanoparticles: The good solvent case,” The Journal of Chemical Physics 135, 214902 (2011).
- Meng et al. (2012) Dong Meng, Sanat K Kumar, J Matthew D Lane, and Gary S Grest, “Effective interactions between grafted nanoparticles in a polymer matrix,” Soft Matter 8, 5002–5010 (2012).
- Milner et al. (1988) Scott T Milner, Thomas A Witten, and Michael E Cates, “Theory of the grafted polymer brush,” Macromolecules 21, 2610–2619 (1988).
- Milner (1991) Scott Thomas Milner, “Polymer brushes,” Science 251, 905–914 (1991).
- Centeno et al. (2014) R Catarino Centeno, E Pérez, and A Gama Goicochea, “On the potential of mean force of a sterically stabilized dispersion,” Journal of Coatings Technology and Research 11, 1023–1031 (2014).
- Johnson (1987) Kenneth Langstreth Johnson, Contact Mechanics (Cambridge University Press, Cambridge, 1987).
- Koh et al. (2020) Clement Koh, Gary S Grest, and Sanat K Kumar, “Assembly of polymer-grafted nanoparticles in polymer matrices,” ACS Nano 14, 13491–13499 (2020).
- Milchev et al. (2008) A Milchev, DI Dimitrov, and K Binder, “Excess free energy of nanoparticles in a polymer brush,” Polymer 49, 3611–3618 (2008).