Effects of linker flexibility on phase behavior and structure of linked colloidal gels
Abstract
Colloidal nanocrystal gels can be assembled using a difunctional “linker” molecule to mediate bonding between nanocrystals. The conditions for gelation and the structure of the gel are controlled macroscopically by the linker concentration and microscopically by the linker’s molecular characteristics. Here, we demonstrate using a toy model for a colloid–linker mixture that linker flexibility plays a key role in determining both phase behavior and structure of the mixture. We fix the linker length and systematically vary its bending stiffness to span the flexible, semiflexible, and rigid regimes. At fixed linker concentration, flexible-linker and rigid-linker mixtures phase separate at low colloid volume fractions in agreement with predictions of first-order thermodynamic perturbation theory, but the semiflexible-linker mixtures do not. We correlate and attribute this qualitatively different behavior to undesirable “loop” linking motifs that are predicted to be more prevalent for linkers with end-to-end distances commensurate with the locations of chemical bonding sites on the colloids. Linker flexibility also influences the spacing between linked colloids, suggesting strategies to design gels with desired phase behavior, structure, and by extension, structure-dependent properties.
I Introduction
Inorganic nanocrystals (NCs) exhibit distinctive optoelectronic properties such as quantum dot emission, localized surface plasmon resonance (LSPR), and electrocatalytic activity that are controlled by their size, shape, and composition [1, 2, 3, 4, 5]. Properties of individual NCs can be further modified in assembled colloidal structures through coupling to neighboring NCs [6]. Coupling effects, such as shifts in the LSPR and enhancement of the local electric field, are sensitive to the distance between (and number of) adjacent NCs [4, 5], so structure gives another powerful route to tune properties of materials made from NCs. Ordered NC structures like superlattices have long been sought [7] but only a limited number of (typically close-packed) lattices have been realized [8]. Gels, i.e., disordered solid-like networks of NCs, are interesting alternative assemblies because their open fractal structures can exhibit varying degrees of NC connectivity giving rise to tunable mechanical or optical properties [9, 10, 11]. NC gels have been proposed as photovoltaic materials and electrocatalysts [12, 13, 14], and the low NC density in gels also makes them attractive candidates for dynamically reconfigurable materials [15, 16, 17, 18, 19].
There are various routes to NC gelation [20, 21], but we have recently demonstrated one controllable, reversible strategy based on dynamic covalent chemistry [22]. Metal oxide NCs were functionalized with ligands bearing an aldehyde group that could react with a complementary hydrazide group on a difunctional “linker” molecule. In qualitative agreement with theoretical predictions [23, 24], gelation occurred upon addition of a sufficient amount of linker and could be subsequently reversed upon its dilution. Linker-mediated gelation is experimentally convenient because it is macroscopically controlled by the linker concentration rather than microscopically controlled by the NC functionality. Furthermore, the large synthetic toolbox of organic chemistry offers significant tunability via molecular design of the ligands or linkers. For example, the length (molecular weight) of the ligand–linker complex not only constrains the allowable spacing between two linked NCs but also affects the phase behavior of the NC–linker mixture. We have shown for a model NC–linker system that, at fixed linker-to-NC number ratio, longer linkers compress the unstable region of the phase diagram (where gels can form by arrested spinodal decomposition upon quenching) to lower NC volume fractions [24].
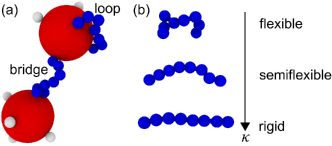
Internal molecular degrees of freedom, such as bending stiffness, also play an important role in linker-mediated gelation. We have found that flexible ligands (linkers) can form “self-links” or “loops,” with difunctional linkers bonding both of their ends to ligands on the same NC (Figure 1a). When present, such bonding motifs tend to inhibit gelation [24, 22]. Similar behavior was recently reported in hydrogels made from trivalent DNA nanostars linked by difunctional double-stranded DNA [26]. Double-stranded DNA is a stiff molecule with a relatively large persistence length, but flexibility can be added to a short DNA linker by including single-stranded DNA joints near its ends [27]. Double-stranded DNA linkers with flexible joints produced a cluster fluid instead of the expected gel because the linker was able to form a loop with two arms of the same nanostar [26].
Synthetic ligands and linkers can be designed to have chemical structures that span the flexible, semiflexible, and rigid regimes of bending stiffness (Figure 1b). Here we highlight a few potential synthetic strategies for realizing such molecules. Flexible molecules, e.g., alkyl chains, are readily made by a variety of techniques; in Ref. 22, we designed and synthesized a flexible ligand for our NCs under amide coupling conditions that are commonly used for preparing natural and unnatural polypeptides [28, 29]. At the other extreme, rigid (rodlike) molecules can be realized using aromatic heterocycles with extended delocalization of -elections; for example, poly[benzol,2-d:5,4-d’bisoxazole-2,6-diyl)-1,4-phenylene] (PBO) and poly[benzo 1,2-d:4,5-d’bisthiazole-2,6-diyl)-1,4-phenylene] (PBZT) are rigid polymers consisting of conjugated benzobisazole and benzobisthiazole rings, respectively. Rigid polymers can be prepared under polymerization condensation conditions [30, 31], but bonds between aryl rings will also form under copper-, nickel-, or palladium-catalyzed cross-coupling conditions,[32, 33, 34, 35] including Kumada [36], Negishi [37], Stille [38], and Suzuki [39] coupling schemes. Semiflexible molecules, with stiffness intermediate between flexible and rigid molecules, can be produced by introducing flexible alkyl chains like polyethylene glycol between rigid, planar aryl moieties under similar synthetic conditions used for rigid molecules [40, 41]. Techniques such as Hiyama-coupling conditions [42] can also incorporate methylene groups between aryl motifs [43, 44, 45, 46, 47, 48]. The length of the flexible alkyl chains or number of methylene groups between aryl motifs can be used to make the molecule more flexible or rigid.
The large space of potential ligand and linker designs with varying molecular weight and flexibility promises significant tunability, but little is known theoretically about how flexibility might affect the phase behavior and structure of linker-mediated NC gel assemblies. To address this gap, here we perform a fundamental study using a toy colloid–linker model (Section II). Building on recent work [24], we systematically introduce bending stiffness to the linkers and show (Section III) that the phase behavior is highly sensitive to the number of loops that form; phase separation (gelation) is suppressed by large loop fractions at fixed colloid and linker concentrations. Linker flexibility also systematically alters the gel structure (and by extension structure-influenced properties), suggesting further avenues for engineering linker-mediated NC gelation (Section IV).
II Model and Simulation Methods
As in our previous work [24], we studied a colloid–linker mixture having colloids and linkers in a volume at temperature . To focus on the effects of flexibility, we considered linkers that were all linear chains having beads of diameter and mass . The colloids were modeled as larger beads with diameter and mass . The linker-to-colloid number ratio of the mixture was set to , and different values of the colloid volume fraction were considered.
All particles were treated as nearly hard spheres interacting through the core-shifted Weeks-Chandler-Andersen potential [49],
| (1) |
where , is Boltzmann’s constant, is the distance between the centers of two particles of types and , and shifts the divergence of the potential to account for the diameters, and , of each particle type. The cutoff for the potential was chosen as to give a purely repulsive interaction.
Within a linker, pairs of bonded beads were additionally joined by finitely extensible nonlinear elastic springs [50],
| (2) |
Standard parameter choices and were adopted [51], and the average linker bond length was . The linker flexibility was controlled by including a bending potential between sets of three consecutively bonded beads [52, 53],
| (3) |
where is the angle between the beads and sets the bending stiffness. For , the persistence length can be estimated as [52], which should be compared to the average contour length . The linker is considered flexible when , semiflexible when , and rigid when . We studied bending stiffnesses to span these regimes, giving .
Chemical bonding between functional end groups on the linkers with specific bonding sites on the colloids was modeled by fixing six beads (each having nominal mass and diameter ) on the surface of each colloid at radius . The end beads of the linkers had a short-ranged attraction to the colloid bonding sites,
| (4) |
where sets the bond strength. The large limit corresponds to potential experimental realizations of our model using, e.g., dynamic covalent chemistry to form strong but reversible bonds [54, 55, 56, 22]. Consistent with this motivation, the colloid bonding sites also repelled each other via Eq. (1) to ensure that only one site could bond with one linker bead at a time. The sites were initially positioned at the vertices of an octahedron, but we also investigated the effects of other arrangements (Section III.3).
We performed classical molecular dynamics simulations of our model using lammps (22 Aug 2018) [57]. The integration timestep was , where is the unit of time, and constant temperature was maintained using a Langevin thermostat with friction coefficient applied to each linker bead and colloid. Random initial configurations of colloids and linkers were prepared in cubic simulation boxes with periodic boundary conditions. The colloid volume fraction was varied from to focus on the dilute conditions typically relevant for preparing low-density nanocrystal gels [9, 10, 11, 12, 13, 14]. For a given bending stiffness , the configurations were first equilibrated for without any attraction (). Next, the attraction was slowly switched on to over a period of using a linear ramp, followed by a equilibration period. We then linearly ramped the attraction to over a period of and held it constant for . We saved the configuration from the end of this period and repeated the incrementing process until . Finally, production simulations of were performed at each and with configurations sampled every for analysis.
In order to determine approximate phase boundaries for the colloid–linker mixture, which give guidance about conditions that may lead to gelation, we computed the partial static structure factor of the colloids [58],
| (5) |
Here is a wavevector in the cubic box of volume , is a vector of integers, and is the position of the th colloid. The angle brackets indicate an ensemble average, which was computed as a time average. We determined the average for the 22 smallest nonzero wavevector magnitudes and extrapolated to by fitting a Lorentzian form [59],
| (6) |
where is a correlation length. We operationally defined structures having to be phase separated [60, 23, 24].
III Results and Discussion
III.1 Phase behavior
The phase behavior of a dilute mixture of colloids and semiflexible or rigid linkers is expected to be qualitatively similar to that of mixtures of patchy colloids [23] or colloids and flexible linkers [24]. The homogeneous colloid–linker mixture is typically thermodynamically stable when the attraction between the colloids and linkers is weak (small ) but can become unstable to composition fluctuations when the attraction is strong (large ) and enough linker is added (increasing ), leading to separation into colloid-rich and colloid-lean phases. We note that the stable fluid phase can be reentrant with respect to [23, 24], but our simulations are performed in the linker-limited regime where increasing destabilizes the fluid.
The thermodynamically unstable regions of the phase diagram are demarcated by the spinodal boundary [58, 61]. Mixtures prepared inside this region will spontaneously phase separate by spinodal decomposition, and this process typically produces kinetically arrested gels [20]. Mixtures prepared outside both the spinodal region and the binodal region (demarcating true phase coexistence) remain single phase but can form gels if the lifetime of the linker-mediated connections between colloids becomes long compared to the observation time [62, 20]. Because of their inherent thermodynamic stability, these “equilibrium gels” are more resistant to aging than gels formed by arrested spinodal decomposition. Theoretical knowledge of the phase diagram can guide design of materials that controllably gel by one these routes.
We previously applied Wertheim’s first-order thermodynamic perturbation theory (TPT) [63, 64, 65, 66, 67] to predict the conditions for phase separation in a mixture of colloids and fully flexible () linkers [24]. Within TPT, the Helmholtz free-energy density is decomposed into two contributions, . is the free-energy density of a reference system without bonding between components, and is the free-energy density due to bonding. For fully flexible linkers, the hard-chain fluid [67] is a useful reference system and has
| (7) |
The first term is the free-energy density of an ideal gas of chain molecules,
| (8) |
where the sum is taken over the different components , each of which consists of chains of beads of equal diameter . Here, is the number density, and is the thermal wavelength of component . For our model, there are two components: colloids each having bead of diameter and with , and linkers having beads of diameter and with . The second term in Eq. (7), , is the excess free-energy density of the hard-sphere fluid obtained by dissociating all bonds from the chains [68, 69],
| (9) |
where . The third term is the excess free-energy density of chain formation [67], where is the contact value of the radial distribution function for two beads of component in the hard-sphere fluid [68]. Note that Eq. (7) corrects a typographical error in Eq. (5) of Ref. 24, where the ideal gas and excess hard-sphere free energies were incorrectly described; Ref. 24 in fact used Eq. (7) to perform calculations.
To compute the bonding free-energy density , it is usually assumed that to first order each site can bond with at most one other site, that bonding is uncorrelated between sites, and that only treelike bonded networks form [70]. For a mixture of components each having identical bonding sites, can be expressed as [67]
| (10) |
where is the fraction of sites not bonded on component . In our model, the colloids have sites and the linkers have sites. The values of are determined by the chemical equilibrium equations,
| (11) |
where only is nonzero for our model and is given by [71]
| (12) |
Here, is the vector from the center of the colloid to the end of the linker participating in the bond, is the vector of Euler angles defining the orientation of the colloid, and is the end-to-end vector of the linker. The integrand is the product of the distribution of end-to-end vectors in the hard-chain reference fluid, the colloid–linker pair correlation function in the hard-chain reference fluid, and the Mayer -function for a designated bonding site on the colloid and the end of the linker at . The -function, and hence the integrand, is nonzero only when the two sites interact, which occurs over a short range given the form of Eq. (4). can be regarded as a “bond volume” averaged over orientations of the colloid and conformations of the linker. We evaluated using the same methodology as in Ref. 71 [72, 73, 74], where and were obtained from simulations of the hard-chain reference fluid; complete details are given in the Supporting Information.
Given the Helmholtz free energy , the spinodal boundary can be computed using the matrix of second derivatives with respect to density [61], where . The limit of stability occurs when the determinant of is zero. For the fully flexible linkers (), the TPT predictions are in good agreement with the simulations (Figure 2a). (The TPT predictions are also improved compared to Ref. 24 because Eq. (12) uses a more accurate approximation of the pair correlation function.) Phase separation was mainly observed in the simulations inside the TPT spinodal; Figure 2b shows a representative phase-separated morphology at and . We previously investigated how the phase behavior of mixtures of colloids and fully flexible linkers depended on the linker length [24], so here we focus on the role of flexibility.
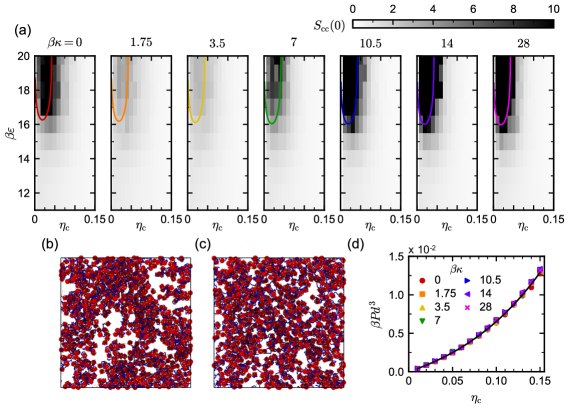
As a perturbation theory, the TPT free energy is sensitive to the reference fluid model, i.e., without linking. In particular, the free-energy density depends on both the reference fluid’s equation of state, which determines , and its structure, which enters through . We hence expect similar phase behavior for different bending stiffnesses if the equation of state and relevant structures of the reference fluid do not depend strongly on . We performed simulations of mixtures having , , and varied and , and we measured the pressure (Figure 2d), the distribution of linker end-to-end vectors (Figure 3), and the pair distribution function (Figures S1 and S2). The pressure changed negligibly with and was fully consistent with the pressure derived from Eq. (7) [67]. The end-to-end vector distribution changed significantly with , shifting its peak to larger for larger as expected. The pair distribution function was again far less sensitive to . It tended to be less than 1 for conformations relevant to evaluating Eq. (12) due to depletion of the polymer-like chains near the surface of the colloid [75], with slightly larger values for the stiffer linkers. Overall, though, the change in at was only 30% between and .
Given this analysis of the reference fluid, we expected similar phase behavior at all values of . Indeed, for (the rigid linkers), the simulated phase diagrams were essentially the same, closely resembling that at and the TPT predictions. Surprisingly, though, the semiflexible linkers () exhibited very different behavior than predicted by first-order TPT. For and , only minor structuring was detected at all and , with well below the threshold we considered phase separated. This difference was visually apparent in simulation snapshots taken at conditions inside the TPT spinodal. Unlike when (Figure 2b), significant colloid density variations were not visible at and when (Figure 2c). The lack of phase separation for only the semiflexible linkers could not be explained using first-order TPT.
III.2 Linker loops
We suspected that the striking discrepancy between the simulations and TPT for the semiflexible linkers was caused by certain linking motifs that are prevalent in the simulations at these flexibilities but are not accounted for in first-order TPT. In previous work, we have speculated that the presence of cycles within the graph representing the bonded network of colloids and linkers might suppress phase separation [24, 22]. In particular, we hypothesized that for a fixed total amount of linker the fluid phase might be stabilized if a substantial number of linkers formed “loops” by attaching both ends to the same colloid, rather than forming “bridges” between two different colloids; a similar explanation was recently proposed for the lack of gelation in DNA hydrogels linked with flexible joints [26]. This definition of a loop, which can also be interpreted as a “double bond” between a linker and a colloid, is the smallest cycle that can form, and other cycles like double links between two colloids are included with the bridges here.
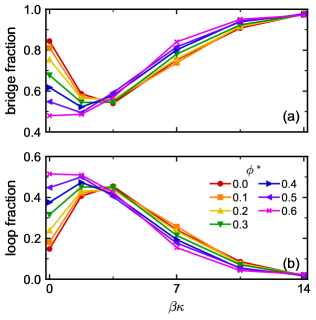
We counted the number of linkers forming either bridges or loops in our simulations (Figure 4), focusing on the strong attraction regime () where essentially all linkers had both ends bonded in one of these motifs. For the fully flexible linkers, a small fraction of linkers formed loops (Figure 4b) rather than bridges (Figure 4a) at all , with the loop fraction tending to be smaller at larger . An overwhelming majority of rigid linkers formed bridges at all , constituting of the links for . Interestingly, and consistent with our hypothesis, there was a pronounced increase in the loop fraction (Figure 4b) for the semiflexible linkers, which had a maximum at roughly . At , nearly 50% of the linkers formed loops when and the mixture remained essentially single phase, despite phase separating for both the fully flexible and rigid linkers (Figure 2a).
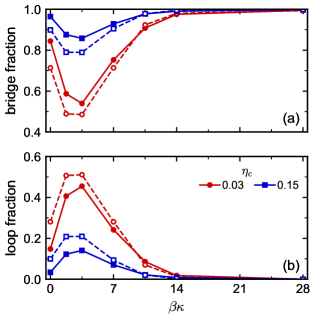
We recently extended first-order TPT to include linker loops (double bonds) [71], and this theory readily predicts the fraction of linkers forming bridges or loops in a spatially homogeneous fluid at equilibrium. We computed these fractions at and for all chain flexibilities using a similar methodology as in Ref. 71; numerical details are given in the Supplementary Material. The TPT predictions for the fraction of linkers forming loops are in excellent agreement with the simulations (Figure 4), supporting the notions that (1) loops are equilibrium bonding motifs and (2) for the studied colloid bonding site geometry and linker length, loops are more prevalent at intermediate flexibilities. We note that the TPT neglects any phase separation in the simulations at ; this does not seem to qualitatively affect the result.
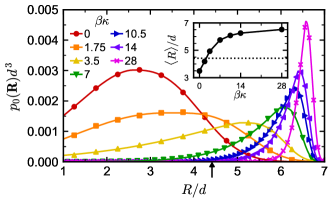
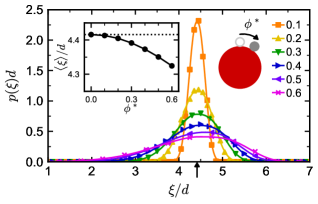
The maximum in loop fraction with respect to can be understood using the distribution of linker end-to-end vectors (Figure 3). In order to form a loop, both ends of the linker must attach to sites on the same colloid, so loops become more prevalent when it is more probable for the linker to have an end-to-end distance commensurate with the colloid site–site distance. We consider only loops between patches at neighboring vertices of the octahedron, which are separated by distance (arrow in Figure 3), because loops between vertices on opposite hemispheres of the colloid are forbidden by the linker contour length. As increases, this end-to-end distance initially becomes more probable but then becomes significantly less likely once the linkers become rigid. The average end-to-end distance (inset of Figure 3) is also comparable to the site–site distance (dotted line) in the semiflexible regime where the most loops are observed. Qualitatively, the flexible linkers pay an entropic penalty to stretch beyond their preferred size and form a loop, while the rigid linkers pay a bending-energy penalty to do the same; in contrast, the semiflexible linkers are naturally compatible with the length scale required to form loops in our model. These effects of flexibility measured by manifest in the TPT as an increase in the double-bond volume [71], which is nearly three times larger when than when .
III.3 Colloid bonding site geometry
An immediate consequence of this interpretation is that the loop fraction should also depend on the distribution of distances between bonding sites on the colloids. A distribution that overlaps significantly with should favor loop formation, while an incompatible should produce fewer loops. The impact of the colloid bonding site geometry on loop formation is not only of theoretical interest but also a practical consideration for experiments, where might be determined by the details of the surface functionalization and polydispersity in surface characteristics is inherent to synthesis. To test the sensitivity of properties to the bonding site geometry, we randomly displaced the colloid bonding sites from their initial positions at the vertices of an octahedron. As illustrated in Figure 5, each site was pertubed by a uniformly random amount on the surface of the sphere with radius up to a maximum polar angle . Adjusting varies from a perfectly monodisperse distribution at to a broad distribution at (Figure 5), which is close to the maximum polar angle that still guarantees two sites will not overlap. (We include in only the distances between sites that were initially nearest neighbors on the octahedron, as these are the ones most likely to form loops even after being displaced.) Generating bonding site polydispersity in this way ensures that the average site–site distance remains nearly constant (decreasing only 2% over the entire range) and allows us to isolate the effects of site polydispersity from effects of the average site–site distance.
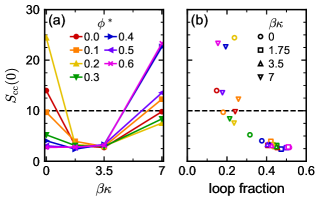
We simulated the colloid–linker mixture with polydisperse bonding sites at , which is inside the first-order TPT spinodal and led to phase separation for the flexible and rigid linkers. We will again focus on the loop fraction and phase behavior in the strong attraction limit . For , the bridge and loop fractions were nearly unchanged from the monodisperse () case, with the loop fraction having a maximum for the semiflexible linkers (Figure 6). Further increase in significantly increased the loop fraction for the flexible linkers, but the rigid linkers were less affected. It is sensible that increasing had a more dramatic effect on the flexible linkers than the rigid linkers because for the flexible linkers is broad (Figure 3); perturbing the colloid bonding sites brings some sites closer together (Figure 5) where there are probable flexible-linker conformations that previously did not produce loops. For the rigid linkers, increasing did little to increase the overlap between the two distributions, and the loop fraction correspondingly changed little with .
We also measured the colloid partial structure factor (Figure 7a) to interrogate how the thermodynamic stability of the fluid phase depended on . We show only the results for because all rigid-linker mixtures phase separated regardless of . Based on the measured loop fractions, we expected and observed similar phase behavior when as for the monodisperse () bonding site geometry, with consistent with a lack of phase separation in the semiflexible-linker mixtures. For larger , phase separation was also suppressed for the flexible linkers, which had a significant increase in the loop fraction. Indeed, we found a strong negative correlation between and the loop fraction (Figure 7b); indicated phase separation when less than 30% of linkers formed loops. This strongly suggests that controlling the loop fraction, e.g., by designing the linker length or flexibility, is a key component of engineering the phase behavior of the colloid–linker mixture and the conditions that may lead to gelation.
III.4 Structure
So far, we have focused our discussion on controlling phase behavior, but in our previous work with flexible linkers, we showed that adjusting the linker length also gave control over the microstructure of the assembled colloids [24]. Longer linkers having larger tended to increase the spacing between colloids, which may have important consequences for the optical properties of gels containing plasmonic nanocrystals [4, 5]. Linker flexibility gives another potential handle to control the colloid microstructure because semiflexible or rigid linkers have larger than flexible linkers of equal contour length (Figure 3).
We measured the colloid–colloid pair correlation function (Figure S3) for various compositions and flexibilities in the strong attraction limit (). We focused on the position of the maximum in (Figure 8a), which identifies the typical distance to a colloid’s nearest neighbor. At small , shifted to larger distances with increasing , which is consistent with the corresponding increase in . Interestingly, this shift did not depend on whether the mixture phase separated, and was nearly constant for a given when . At larger , decreased with increasing and ultimately reached nearly the same value for all at . The flexible linker seems to be an exception, having nearly constant over the entire range of we simulated.
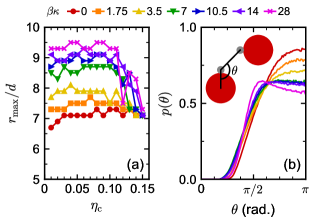
Due to many-body effects in the linked-colloid assemblies, the measured cannot be simply explained using only the average end-to-end size of the linker . Assuming a colinear arrangement of two colloids and a linker, we might expect , which is and for and , respectively; both are considerably larger than the measured . To obtain a smaller , the linkers must either compress or connect two colloids at an angle with each other. A significant decrease in the end-to-end distance of the rigid linkers would incur a large bending penalty and is unlikely, so we accordingly measured the bond angle between pairs of linked colloids (Figure 8b). We defined using the center and bonded patch of the first colloid and the bonded patch on the second colloid. The distribution of bond angles showed predominantly colinear arrangements () for the flexible linkers, but as increased, the bond angles were forced toward more right-angle () arrangements. This change in allows the colloids to pack closely without compressing the linker and seems to supress formation of a single dominant length scale in the assemblies, producing a more uniform distribution of pair distances (Figure S3).
IV Conclusions
We have shown that flexibility can play an important role in determining the phase behavior and structure of linked colloidal assemblies. In our simulations of a toy model for a colloid–linker mixture, phase separation, which can lead to gelation, was suppressed when the linker’s internal degrees of freedom allowed it to readily attach both of its ends to the same colloid in a “loop.” This finding is fully consistent with recent experiments with trivalent DNA nanostars [26], despite the significantly higher number of potential bonding sites on the colloids we studied. The linker loop fraction can be predicted theoretically using an extension of thermodynamic perturbation theory [71], and more efficient linking may then be obtained by designing linker molecules with size, flexibility, or functionality that disfavor or disallow loop motifs. Linker flexibility also gives a handle for controlling the microstructure of the assembled colloids, particularly at small ; however, changes in the size of the linker with flexibility do not straightforwardly propagate to the typical colloid–colloid separation, especially at larger . Since many nanoparticle gels are prepared at relatively low volume fractions, flexibility may still be a viable strategy for controlling both gel microstructure and phase behavior. Assembly schemes that use mixtures of linkers having different lengths as well as different flexibilities are especially intriguing, as they may give independent tunability of phase behavior and microstructure [24].
Supplementary Material
See supplementary material for details of the TPT calculations including loops, simulated reference fluid data, and colloid–colloid pair correlation functions in linked assemblies.
Acknowledgements.
This research was primarily supported by the National Science Foundation through the Center for Dynamics and Control of Materials: an NSF MRSEC under Cooperative Agreement No. DMR-1720595, with additional support from an Arnold O. Beckman Postdoctoral Fellowship (ZMS), a National Science Foundation Graduate Research Fellowship under Grant No. DGE-1610403 (SAV), and the Welch Foundation (Grant Nos. F-1696 and F-1848). We acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources.Data Availability
The data that support the findings of this study are available from the authors upon reasonable request.
References
References
- El-Sayed [2001] M. A. El-Sayed, “Some Interesting Properties of Metals Confined in Time and Nanometer Space of Different Shapes,” Acc. Chem. Res. 34, 257–264 (2001).
- El-Sayed [2004] M. A. El-Sayed, “Small is different: Shape-, size-, and composition-dependent properties of some colloidal semiconductor nanocrystals,” Acc. Chem. Res. 37, 326–333 (2004).
- Yin and Alivisatos [2005] Y. Yin and A. P. Alivisatos, “Colloidal nanocrystal synthesis and the organic–inorganic interface,” Nature 437, 664–670 (2005).
- Talapin et al. [2010] D. V. Talapin, J.-S. Lee, M. V. Kovalenko, and E. V. Shevchenko, “Prospects of Colloidal Nanocrystals for Electronic and Optoelectronic Applications,” Chem. Rev. 110, 389–458 (2010).
- Agrawal et al. [2018] A. Agrawal, S. H. Cho, O. Zandi, S. Ghosh, R. W. Johns, and D. J. Milliron, “Localized Surface Plasmon Resonance in Semiconductor Nanocrystals,” Chem. Rev. 118, 3121–3207 (2018).
- Boles, Engel, and Talapin [2016] M. A. Boles, M. Engel, and D. V. Talapin, “Self-Assembly of Colloidal Nanocrystals: From Intricate Structures to Functional Materials,” Chem. Rev. 116, 11220–11289 (2016).
- Collier, Vossmeyer, and Heath [1998] C. P. Collier, T. Vossmeyer, and J. R. Heath, “Nanocrystal Superlattices,” Annu. Rev. Phys. Chem. 49, 371–404 (1998).
- Murray, Kagan, and Bawendi [2000] C. B. Murray, C. R. Kagan, and M. G. Bawendi, “Synthesis and Characterization of Monodisperse Nanocrystals and Close-Packed Nanocrystal Assemblies,” Annu. Rev. Mater. Sci. 30, 545–610 (2000).
- Arachchige and Brock [2007] I. U. Arachchige and S. L. Brock, “Sol–Gel Methods for the Assembly of Metal Chalcogenide Quantum Dots,” Acc. Chem. Res. 40, 801–809 (2007).
- Ziegler et al. [2017] C. Ziegler, A. Wolf, W. Liu, A.-K. Herrmann, N. Gaponik, and A. Eychmüller, “Modern Inorganic Aerogels,” Angew. Chem. Int. Ed. 56, 13200–13221 (2017).
- Rechberger and Niederberger [2017] F. Rechberger and M. Niederberger, “Synthesis of Aerogels: From Molecular Routes to 3-Dimensional Assembly,” Nanoscale Horiz. 2, 6–30 (2017).
- Nozik [2010] A. J. Nozik, “Semiconductor Quantum Dots and Quantum Dot Arrays and Applications of Multiple Exciton Generation to Third-Generation Photovoltaic Solar Cells,” Chem. Rev. 110, 6873–6890 (2010).
- Gaponik, Herrmann, and Eychmüller [2011] N. Gaponik, A.-K. Herrmann, and A. Eychmüller, “Colloidal Nanocrystal-Based Gels and Aerogels: Material Aspects and Application Perspectives,” J. Phys. Chem. Lett. 3, 8–17 (2011).
- Cai et al. [2018] B. Cai, V. Sayevich, N. Gaponik, and A. Eychmüller, “Emerging Hierarchical Aerogels: Self-Assembly of Metal and Semiconductor Nanocrystals,” Adv. Mater. 30, 1707518 (2018).
- Lesnyak et al. [2011] V. Lesnyak, A. Wolf, A. Dubavik, L. Borchardt, S. V. Voitekhovich, N. Gaponik, S. Kaskel, and A. Eychmüller, “3D Assembly of Semiconductor and Metal Nanocrystals: Hybrid CdTe/Au Structures with Controlled Content,” J. Am. Chem. Soc. 133, 13413–13420 (2011).
- Borsley and Kay [2016] S. Borsley and E. R. Kay, “Dynamic Covalent Assembly and Disassembly of Nanoparticle Aggregates,” Chem. Commun. 52, 9117–9120 (2016).
- Saez Cabezas et al. [2018] C. A. Saez Cabezas, G. K. Ong, R. B. Jadrich, B. A. Lindquist, A. Agrawal, T. M. Truskett, and D. J. Milliron, “Gelation of Plasmonic Metal Oxide Nanocrystals by Polymer-Induced Depletion Attractions,” Proc. Natl. Acad. Sci. 115, 8925–8930 (2018).
- Wang et al. [2019] Y. Wang, P. J. Santos, J. M. Kubiak, X. Guo, M. S. Lee, and R. J. Macfarlane, “Multistimuli Responsive Nanocomposite Tectons for Pathway Dependent Self-Assembly and Acceleration of Covalent Bond Formation,” J. Am. Chem. Soc. 141, 13234–13243 (2019).
- Marro, della Sala, and Kay [2020] N. Marro, F. della Sala, and E. R. Kay, “Programmable Dynamic Covalent Nanoparticle Building Blocks with Complementary Reactivity,” Chem. Sci. 11, 372–383 (2020).
- Zaccarelli [2007] E. Zaccarelli, “Colloidal Gels: Equilibrium and Nonequilibrium Routes,” J. Phys.: Condens. Matter 19, 323101 (2007).
- Matter, Luna, and Niederberger [2020] F. Matter, A. L. Luna, and M. Niederberger, “From Colloidal Dispersions to Aerogels: How to Master Nanoparticle Gelation,” Nano Today 30, 100827 (2020).
- Dominguez et al. [2020] M. N. Dominguez, M. P. Howard, J. M. Maier, S. Valenzuela, Z. M. Sherman, L. C. Reimnitz, J. Kang, S. H. Cho, S. L. Gibbs, A. K. Menta, D. L. Zhuang, A. van der Stok, S. J. Kline, E. V. Anslyn, T. M. Truskett, and D. J. Milliron, “Assembly of Linked Nanocrystal Colloids by Reversible Covalent Bonds,” Chem. Mater. (2020), 10.1021/acs.chemmater.0c04151.
- Lindquist et al. [2016] B. A. Lindquist, R. B. Jadrich, D. J. Milliron, and T. M. Truskett, “On the formation of equilibrium gels via a macroscopic bond limitation,” J. Chem. Phys. 145, 074906 (2016).
- Howard et al. [2019] M. P. Howard, R. B. Jadrich, B. A. Lindquist, F. Khabaz, R. T. Bonnecaze, D. J. Milliron, and T. M. Truskett, “Structure and phase behavior of polymer-linked colloidal gels,” J. Chem. Phys. 151, 124901 (2019).
- Stukowski [2010] A. Stukowski, “Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool,” Modell. Simul. Mater. Sci. Eng. 18, 015012 (2010).
- Stoev et al. [2020] I. D. Stoev, T. Cao, A. Caciagli, J. Yu, C. Ness, R. Liu, R. Ghosh, T. O’Neill, D. Liu, and E. Eiser, “On the role of flexibility in linker-mediated DNA hydrogels,” Soft Matter 16, 990–1001 (2020).
- Xing et al. [2018] Z. Xing, A. Caciagli, T. Cao, I. Stoev, M. Zupkauskas, T. O’Neill, T. Wenzel, R. Lamboll, D. Liu, and E. Eiser, “Microrheology of DNA hydrogels,” Proc. Nat. Acad. Sci. USA 115, 8137–8142 (2018).
- Chen, Zaro, and Shen [2013] X. Chen, J. L. Zaro, and W.-C. Shen, “Fusion protein linkers: Property, design and functionality,” Adv. Drug Delivery Rev. 65, 1357–1369 (2013).
- Wriggers, Chakravarty, and Jennings [2005] W. Wriggers, S. Chakravarty, and P. A. Jennings, “Control of protein functional dynamics by peptide linkers,” Pept. Sci. 80, 736–746 (2005).
- Wolfe and Arnold [1981] J. F. Wolfe and F. E. Arnold, “Rigid-rod polymers. 1. Synthesis and thermal properties of para-aromatic polymers with 2,6-benzobisoxazole units in the main chain,” Macromolecules 14, 909–915 (1981).
- Hu et al. [2003] X.-D. Hu, S. E. Jenkins, B. G. Min, M. B. Polk, and S. Kumar, “Rigid‐Rod Polymers: Synthesis, Processing, Simulation, Structure, and Properties,” Macromol. Mater. Eng. 288, 823–843 (2003).
- Ullmann and Bielecki [1901] F. Ullmann and J. Bielecki, “Ueber Synthesen in der Biphenylreihe,” Ber. Dtsch. Chem. Ges. 34, 2174–2185 (1901).
- Percec, Bae, and Hill [1995] V. Percec, J.-Y. Bae, and D. H. Hill, “Aryl Mesylates in Metal Catalyzed Homo- and Cross-Coupling Reactions. 4. Scope and Limitations of Aryl Mesylates in Nickel Catalyzed Cross-Coupling Reactions,” J. Org. Chem. 60, 6895–6903 (1995).
- Yun et al. [2020] Y.-l. Yun, J. Yang, Y.-h. Miao, J. Sun, and X.-j. Wang, “Recent advances in Palladium(II)-catalyzed activation of aromatic ring C–H bonds,” J. Saudi Chem. Soc. 24, 151–185 (2020).
- Nelson and Crouch [2004] T. D. Nelson and R. D. Crouch, “Cu, Ni, and Pd Mediated Homocoupling Reactions in Biaryl Syntheses: The Ullmann Reaction,” Org. React. 63, 265–555 (2004).
- Tamao, Sumitani, and Kumada [1972] K. Tamao, K. Sumitani, and M. Kumada, “Selective carbon–carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel–phosphine complexes,” J. Am. Chem. Soc. 94, 4374–4376 (1972).
- Negishi and Van Horn [1977] E. Negishi and D. E. Van Horn, “Selective carbon–carbon bond formation via transition metal catalysis. 4. A novel approach to cross-coupling exemplified by the nickel-catalyzed reaction of alkenylzirconium derivatives with aryl halides,” J. Am. Chem. Soc. 99, 3168–3170 (1977).
- McKean et al. [1987] D. R. McKean, G. Parrinello, A. F. Renaldo, and J. K. Stille, “Synthesis of Functionalized Styrenes via Palladium-Catalyzed Coupling of Aryl Bromides with Vinyl Tin Reagents,” J. Org. Chem. 52, 422–424 (1987).
- Miyaura and Suzuki [1995] N. Miyaura and A. Suzuki, “Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds,” Chem. Rev. 95, 2457–2483 (1995).
- Kikutani [2002] T. Kikutani, “Formation and structure of high mechanical performance fibers. II. Flexible polymers,” J. Appl. Poly. Sci. 83, 559–571 (2002).
- Morozova et al. [2018] S. Morozova, P. W. Schmidt, F. S. Bates, and T. P. Lodge, “Effect of Poly(ethylene glycol) Grafting Density on Methylcellulose Fibril Formation,” Macromolecules 51, 9413–9421 (2018).
- Hatanaka and Hiyama [1988] Y. Hatanaka and T. Hiyama, “Pentacoordinate organosilicate as an alkylating reagent: Palladium catalyzed methylation of aryl halides,” Tetrahedron Lett. 29, 97–98 (1988).
- Denmark and Tymonko [2005] S. E. Denmark and S. A. Tymonko, “Sequential Cross-Coupling of 1,4-Bissilylbutadienes: Synthesis of Unsymmetrical 1,4-Disubstituted 1,3-Butadienes,” J. Am. Chem. Soc. 127, 8004–8005 (2005).
- McLaughlin, McAdam, and Cook [2015] M. G. McLaughlin, C. A. McAdam, and M. J. Cook, “MIDA–Vinylsilanes: Selective Cross-Couplings and Applications to the Synthesis of Functionalized Stilbenes,” Org. Lett. 17, 10–13 (2015).
- Miller and Montgomery [2014] Z. D. Miller and J. Montgomery, “Regioselective Allene Hydroarylation via One-Pot Allene Hydrosilylation/Pd-Catalyzed Cross-Coupling,” Org. Lett. 16, 5486–5489 (2014).
- Zhang et al. [2014] P. Zhang, J. Xu, Y. Gao, X. Li, G. Tang, and Y. Zhao, “Synthesis of Diarylmethanes through Palladium-Catalyzed Coupling of Benzylic Phosphates with Arylsilanes,” Synlett 25, 2928–2932 (2014).
- Yoshiaki et al. [2007] N. Yoshiaki, E. Shiro, C. Jinshui, I. Hidekazu, and H. Tamejiro, “Cross-coupling Reaction of Allylic and Benzylic Carbonates with Organo[2-(hydroxymethyl)phenyl]dimethylsilanes,” Chem. Lett. 36, 606–607 (2007).
- Dey, Chattopadhyay, and Ranu [2008] R. Dey, K. Chattopadhyay, and B. C. Ranu, “Palladium(0) Nanoparticle Catalyzed Cross-Coupling of Allyl Acetates and Aryl and Vinyl Siloxanes,” J. Org. Chem. 73, 9461–9464 (2008).
- Weeks, Chandler, and Andersen [1971] J. D. Weeks, D. Chandler, and H. C. Andersen, “Role of Repulsive Forces in Determining Equilibrium Structure of Simple Liquids,” J. Chem. Phys. 54, 5237–5247 (1971).
- Bishop, Kalos, and Frisch [1979] M. Bishop, M. H. Kalos, and H. L. Frisch, “Molecular dynamics of polymeric systems,” J. Chem. Phys. 70, 1299–1304 (1979).
- Grest and Kremer [1986] G. S. Grest and K. Kremer, “Molecular dynamics simulation for polymers in the presence of a heat bath,” Phys. Rev. A 33, 3628–3631 (1986).
- Nikoubashman, Milchev, and Binder [2016] A. Nikoubashman, A. Milchev, and K. Binder, “Dynamics of single semiflexible polymers in dilute solution,” J. Chem. Phys. 145, 234903 (2016).
- Nikoubashman and Howard [2017] A. Nikoubashman and M. P. Howard, “Equilibrium Dynamics and Shear Rheology of Semiflexible Polymers in Solution,” Macromolecules 50, 8279–8289 (2017).
- Rowan et al. [2002] S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders, and J. F. Stoddart, “Dynamic Covalent Chemistry,” Angew. Chem. Int. Ed. 41, 898–952 (2002).
- Jin et al. [2013] Y. Jin, C. Yu, R. J. Denman, and W. Zhang, “Recent Advances in Dynamic Covalent Chemistry,” Chem. Soc. Rev. 42, 6634–6654 (2013).
- Seifert, Trejo, and Anslyn [2016] H. M. Seifert, K. R. Trejo, and E. V. Anslyn, “Four Simultaneously Dynamic Covalent Reactions. Experimental Proof of Orthogonality,” J. Am. Chem. Soc. 138, 10916–10924 (2016).
- Plimpton [1995] S. Plimpton, “Fast Parallel Algorithms for Short-Range Molecular Dynamics,” J. Comput. Phys. 117, 1–19 (1995).
- Hansen and McDonald [2013] J.-P. Hansen and I. R. McDonald, Theory of Simple Liquids with Applications to Soft Matter, 4th ed. (Academic Press, New York, 2013).
- Jadrich et al. [2015] R. B. Jadrich, J. A. Bollinger, K. P. Johnston, and T. M. Truskett, “Origin and detection of microstructural clustering in fluids with spatial-range competitive interactions,” Phys. Rev. E 91, 042312 (2015).
- Zaccarelli et al. [2005] E. Zaccarelli, S. V. Buldyrev, E. La Nave, A. J. Moreno, I. Saika-Voivod, F. Sciortino, and P. Tartaglia, “Model for Reversible Colloidal Gelation,” Phys. Rev. Lett. 94, 218301 (2005).
- Chimowitz [2005] E. H. Chimowitz, Introduction to Critical Phenomena in Fluids (Oxford University Press, New York, 2005).
- Bianchi et al. [2006] E. Bianchi, J. Largo, P. Tartaglia, E. Zaccarelli, and F. Sciortino, “Phase Diagram of Patchy Colloids: Towards Empty Liquids,” Phys. Rev. Lett. 97, 168301 (2006).
- Wertheim [1984a] M. S. Wertheim, “Fluids with Highly Directional Attractive Forces. I. Statistical Thermodynamics,” J. Stat. Phys. 35, 19–34 (1984a).
- Wertheim [1984b] M. S. Wertheim, “Fluids with Highly Directional Attractive Forces. II. Thermodynamic Perturbation Theory and Integral Equations,” J. Stat. Phys. 35, 35–47 (1984b).
- Wertheim [1986a] M. S. Wertheim, “Fluids with Highly Directional Attractive Forces. III. Multiple Attraction Sites,” J. Stat. Phys. 42, 459–476 (1986a).
- Wertheim [1986b] M. S. Wertheim, “Fluids with Highly Directional Attractive Forces. IV. Equilibrium Polymerization,” J. Stat. Phys. 42, 477–492 (1986b).
- Chapman, Jackson, and Gubbins [1988] W. G. Chapman, G. Jackson, and K. E. Gubbins, “Phase equilibria of associating fluids: Chain molecules with multiple bonding sites,” Mol. Phys. 65, 1057–1079 (1988).
- Boublík [1970] T. Boublík, “Hard-Sphere Equation of State,” J. Chem. Phys. 53, 471–472 (1970).
- Howard, Nikoubashman, and Panagiotopoulos [2017] M. P. Howard, A. Nikoubashman, and A. Z. Panagiotopoulos, “Stratification in Drying Polymer–Polymer and Colloid–Polymer Mixtures,” Langmuir 33, 11390–11398 (2017).
- Jackson, Chapman, and Gubbins [1988] G. Jackson, W. G. Chapman, and K. E. Gubbins, “Phase equilibria of associating fluids: Spherical molecules with multiple bonding sites,” Mol. Phys. 65, 1–31 (1988).
- Howard et al. [2020] M. P. Howard, Z. M. Sherman, D. J. Milliron, and T. M. Truskett, “Wertheim’s thermodynamic perturbation theory with double-bond association and its application to colloid–linker mixtures,” arXiv , 2010.08027 (2020).
- Harris et al. [2020] C. R. Harris, K. J. Millman, S. J. van der Walt, R. Gommers, P. Virtanen, D. Cournapeau, E. Wieser, J. Taylor, S. Berg, N. J. Smith, R. Kern, M. Picus, S. Hoyer, M. H. van Kerkwijk, M. Brett, A. Haldane, J. Fernández del Río, M. Wiebe, P. Peterson, P. Gérard-Marchant, K. Sheppard, T. Reddy, W. Weckesser, H. Abbasi, C. Gohlke, and T. E. Oliphant, “Array programming with NumPy,” Nature 585, 357–362 (2020).
- Virtanen et al. [2020] P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa, P. van Mulbregt, and SciPy 1.0 Contributors, “SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python,” Nature Methods 17, 261–272 (2020).
- Lam, Pitrou, and Seibert [2015] S. K. Lam, A. Pitrou, and S. Seibert, “Numba: A LLVM-Based Python JIT Compiler,” in Proceedings of the Second Workshop on the LLVM Compiler Infrastructure in HPC, LLVM ’15 (2015).
- Fuchs and Schweizer [2002] M. Fuchs and K. S. Schweizer, “Structure of colloid–polymer suspensions,” J. Phys.: Condens. Matter 14, R239–R269 (2002).