Charging Dynamics of Electrical Double Layers Inside a Cylindrical Pore: Predicting the Effects of Arbitrary Pore Size
Abstract
Porous electrodes are found in energy storage devices such as supercapacitors and pseudocapacitors. However, the effect of electrode-pore-size distribution over their energy storage properties remains unclear. Here, we develop a model for the charging of electrical double layers inside a cylindrical pore for arbitrary pore size. We assume small applied potentials and perform a regular perturbation analysis to predict the evolution of electrical potential and ion concentrations in both the radial and axial directions. We validate our perturbation model with direct numerical simulations of the Poisson-Nernst-Planck equations, and obtain quantitative agreement between the two approaches for small and moderate potentials. Our analysis yields two main characteristic features of arbitrary pore size: i) a monotonic decrease of the charging timescale with an increase in relative pore size (pore size relative to Debye length); ii) large potential changes for overlapping double layers in a thin transition region, which we approximate mathematically by a jump discontinuity. We quantify the contributions of electromigration and charge diffusion fluxes which provide mechanistic insights into the dependence of charging timescale and capacitance on pore size. We develop a modified transmission circuit model that captures the effect of arbitrary pore size and demonstrate that a time-dependent transition-region resistor needs to be included in the circuit. We also derive phenomenological expressions for average effective capacitance and charging timescale as a function of pore-size distribution. We show that the capacitance and charging timescale increase with smaller average pore sizes and with smaller polydispersity, resulting in a gain of energy density at a constant power density. Overall, our results advance the mechanistic understanding of electrical-double-layer charging.
I Introduction
Batteries and fuel cells are traditional porous-material-based electrochemical devices. Over the past 60 years, new devices such as supercapacitors [1] have started to emerge. They are comprised of porous electrodes – typically made up of dispersions of activated carbon spheres – immersed in aqueous, organic, or ionic liquid electrolytes. The electrodes store charge through the physical adsorption of dissociated ions onto their pore surfaces, forming a charged region commonly referred to as the electrical double layer [2, 3]. Obviating the need for redox reactions, these devices charge faster than batteries and present better cyclability. Nevertheless, their energy density and capacitance are limited by their available specific surface area [3]. In view of these characteristics, supercapacitors bridge the gap between traditional capacitors and batteries, being used in situations where fast response and moderate energy output are required, e.g., stabilization of energy fluctuations in power grids [4] and memory protection in electronic devices [5]. More recently, hybrid capacitors have been designed in an effort to utilize the energy storage mechanisms of both electrical double layers and reduction-oxidation (redox) reactions [6, 7]. They consist of two distinct electrodes, one supercapacitor-like which stores charge physically into double layers, and another metal oxide pseudocapacitor-like which accumulates energy by performing fast oxidation surface reactions [6]. While there have been significant advances in the material design of supercapacitors and hybrid capacitor electrodes, the effect of pore-size distribution on the energy storage properties of these devices remains unclear [6].
The earliest models for electrode charging address the geometry of an electrolyte between flat plates, dating back to the works of Gouy [8], with later contributions from Chapman [9] and Stern [10] to consolidate the well known Gouy-Chapman-Stern model. Notably, a wealth of different effects have been studied to complete this simplified geometrical picture of flat plate charging. Bazant et al. [11] reviewed the previous flat plate charging studies and developed an approach to solve the Poisson-Nernst-Planck (PNP) equations, from the linear regime at low voltages to the nonlinear effects at high voltages. Feicht et al. [12] compared one-dimensional PNP solutions with the experimentally observed reverse peaks in electrolytic cell discharging. Kilic and Bazant [13, 14] derived modified PNP equations including ion-radius effects to study the influence of ion crowding in charge storage. For higher electrolyte concentrations, other effects such as multi-component diffusion given by Stefan-Maxwell fluxes [15], and ion correlations [16, 17, 18] have been studied, for instance in some continuum models [19, 20, 21].
The porous geometry is incorporated in the form of equivalent circuit representations, widely used in the modeling of pore charging in supercapacitor electrodes [22, 23, 24, 25, 26], stemming from the pioneering work of de Levie [27, 28]. Later, Biesheuvel and Bazant [2] extended the circuit to high potentials for capacitive deionization applications. Recent papers [26, 29] further discuss the relationship between the equivalent circuit and the corresponding transmission line continuous equation for pores with finite lengths. Throughout most pore charging models, common assumptions are either of thin double layers [27, 28, 2, 30] within the pores, i.e., such that the length of the charged region is much smaller than the pore size, or overlapping double layers[31, 32], where the charged regions extending from the opposite sides of the surface meet. However, pore sizes can range from less than 2 nm for micropores to more than 50 nm for macropores [6, 25]. Thus, a first-principles approach that extends pore charging models to arbitrary pore sizes is required in order to accurately describe the charging of supercapacitors and predict their properties, such as capacitance, energy density, and power density. Some of the works which address pore-size dependence focus on the equilibrium response [33], while others propose transient transport-equation-based numerical schemes that describe porous network charging for arbitrary pore sizes [34, 35]. However, to the best of our knowledge, the relative importance of charge transport mechanisms for arbitrary pore sizes and their implications on transmission line circuits have not been reported.
In our previous work [36], an analytical model based on the PNP equations was proposed to describe the charging of pores at low applied potentials in the limit of overlapping double layers. Here, inspired by perturbation models on electrokinetics [37, 38, 39, 40, 41, 42], we develop a perturbation expansion model for arbitrary pore sizes – i.e., pore radii – in the limit of small applied potentials. We compare the predictions of the model to direct numerical simulations to show that the perturbation solution yields good results even for moderate applied potentials ( mV). We demonstrate that a modified transmission line circuit (compared to classic supercapacitor literature [27, 28]) includes a resistor representing finite changes in electric potential at a thin entrance region at the mouth of a pore. We derive an effective capacitance to study the effect of pore-size distribution on energy and power densities. Besides addressing transient charging, the solution developed here quantifies the contributions of diffusion and electromigration and provides mechanistic insights into the charging process of pores of arbitrary sizes.
II Problem Formulation
We consider an ideally conducting porous electrode that consists of tortuous pores of different radii, filled with an electrolyte; see the schematic in Fig. 1a. Once a voltage difference is applied, counterions are attracted to the pore surfaces, forming electrical double layers of thicknesses that may be thin or comparable to the pore size [3, 43]. We follow common practice [11, 2, 36] in assuming the formation of a static diffusion layer (SDL), an electroneutral region beside the electrode. Fig. 1b shows a simplified setup with a cylindrical pore of length and radius , where the radial and axial directions are denoted by and , respectively. The SDL, of length and radius , is adjacent to the pore. We denote the cation and anion concentrations by and electric potential by , where is time. The position represents the interface between the pore and the SDL. We assume that the SDL is in contact with the bulk such that and . At , the concentration of ions everywhere is . At , since the electrode material is an ideal conductor, the potential at the surface of the pore . We also assume that the pore surface is ideally blocking, i.e., the flux of ions across the pore surface is zero. In addition, we assume that , , the ions are monovalent, that ion diffusivities inside the pore are equal and given by . The ion diffusivities outside the pore are also assumed equal and are given by . We note that the ion diffusivities inside and outside the pore may be different due to confinement effects. As we show later, the ratio of diffusivities – i.e., – dictates the interaction between the pore and the SDL. We denote Debye length by such that , where is the electrical permittivity. For reference, a 1 M aqueous electrolyte at room temperature has a Debye length nm. The objective of this article is to determine the value of and for an arbitrary relative pore size .

Physically, when the potential is applied on the surface of a pore, oppositely charged ions migrate inside the pore, while the similarly charged ions transport out of the pore. This relative transport of ions produces an electrical current. In addition, due to the charge imbalance, an electrical double layer forms adjacent to the surface. As time progresses, the potential drop across the electrical double layer starts to saturate, ions stop migrating, and the current ceases.
To solve for and , we start by writing the Poisson-Nernst-Planck equations [44, 45, 46]
| (1a) | |||
| (1b) | |||
| where are the ion fluxes. Inside the pore | |||
| (1c) | |||
| and outside the pore | |||
| (1d) | |||
We note that by angular symmetry. We introduce dimensionless charge density , salt concentration , potential , time , axial position , radial position , and gradient operator . Note that . In addition, for (i.e., the pore region), and for (i.e., the static diffusion layer region; see Fig. 1b). With these substitutions, the set of Eqs. (1d) becomes
| (2a) | |||
| (2b) | |||
| (2c) | |||
| where , is the dimensionless charge flux and is the dimensionless salt flux. Inside the pore | |||
| (2d) | |||
| and outside the pore | |||
| (2e) | |||
At , inside the pore, the potential is constant and equal to the wall potential since the electrical double layer hasn’t developed yet. In contrast, the potential is linear in the SDL due to electroneutrality. Therefore, Eqs. (2) are subjected to the following initial conditions
| (3a) | |||
| (3b) | |||
| (3c) | |||
| (3d) | |||
The boundary conditions (BCs) at are given by the bulk condition, or
| (4a) | |||
| (4b) | |||
| (4c) | |||
At the end of the pore, i.e., , the BCs are that gradients of potential and concentration vanish for , implying
| (5) |
Due to the symmetry, the BCs at the center of the system, i.e., , are simply
| (6) |
At the surface of the pore, i.e., and , the BCs are of ideally blocking electrode and constant potential, which are given by
| (7a) | |||
| (7b) | |||
| (7c) | |||
We note that Eq. (7c) represents the ideally conducting electrode BC. Finally, at the boundaries of the SDL, i.e., and , the BCs are vanishing gradients, or
| (8) |
This assumption is valid for non-interacting pores where the radial currents are identically zero, consistent with the treatment of SDL in the literature [2, 36, 29]. We solve the set of Eqs. (2) – (8) numerically using OpenFOAM [47, 48]. The details of geometry, mesh, and algorithm have been described in Ref. [36]. We refer to the solution from OpenFOAM as direct numerical simulations (DNS). Next, we perform a regular perturbation analysis [44] to obtain an analytical expression for and .
III Regular Perturbation Analysis
In this section, we focus on the small potential limit, i.e., , and conduct a regular perturbation analysis to describe the charge and potential inside the pore. To this end, we divide the solution into three regions: (I) SDL, (II) inside the pore, and (III) transition region between the SDL and the mouth of the pore.
III.1 Static Diffusion Layer
The SDL is characterized by electroneutrality, i.e., . Furthermore, for , [2, 36]. Therefore, as per Eq. (2c) and the boundary conditions in Eqs. (4c), (6), and (8), it is straightforward to show that is linear in and is independent of . Mathematically, we write
| (9) |
where , i.e., the centerline potential to the left of the SDL-pore interface, and is to be determined.
III.2 Inside the Pore
The region inside the pore consists of ion concentration and potential varying in time as well as in both radial and axial directions. In the low-potential limit, we propose regular perturbation expansions of the dependent variables in the small parameter , i.e.,
| (10a) | |||
| (10b) | |||
| (10c) | |||
| We also introduce variables and that represent the centerline charge and density. As per our proposed perturbation expansion, | |||
| (10d) | |||
| (10e) | |||
In this article, we only focus on the leading-order and first-order terms.
The leading-order terms, i.e., , and are obtained from the response in the absence of an applied potential, i.e., . Thus, it can be seen that the leading-order coefficients , and satisfy the set of Eqs. (2) with the initial conditions (3) and boundary conditions (5)–(7).
Inserting the expansions provided in Eq. (10) (with , and ) in Eq. (2a) and (2d) and collecting the first-order terms in yields
| (11) |
Similarly, Poisson’s equation – Eq. (1b) – after substitution of the perturbation expansions takes the form
| (12) |
Only the known leading-order term in the salt concentration enters Eq. (11), such that Eqs. (11) and (12) suffice to determine first-order corrections to charge density and potential in the low potential regime. Therefore, we do not need to further solve Eq. (2b).
Since , the diffusion in the radial direction is much faster than in the axial direction and we can assume quasiequilibrium in the radial direction[34]. Thus, with symmetry and ideally blocking conditions (see Eqs. (6) – (7)), we have . Utilizing this condition in Eq. (2d) and collecting the first-order terms in the perturbation expansion, we get
| (13) |
which implies that [36]
| (14) |
where and are to be determined. Next, by utilizing in Eq. (12), we get [36]
| (15) |
Integrating Eq. (15) with and yields
| (16) |
where is the modified Bessel function of the first kind of order . By substituting in Eq. (16), we obtain
| (17) |
Next, by combining Eqs. (16) - (17), substituting the leading- and first-order coefficients in Eq. (10), and neglecting higher-order terms, we get
| (18) |
Similarly, substituting Eqs. (16) and (17) into Eq. (14) and neglecting higher-order terms in the perturbation expansion of , we get
| (19) |
Next, we note that since , so we can simplify Eq. (11) to obtain
| (20) |
To determine the axial dependence, we average Eq. (20) over the cross-sectional area of the pore (integrating across the radial direction) by utilizing the known radial dependence in Eqs. (18) and (19). Mathematically,
| (21) |
We emphasize that this crucial step enables us to derive a solution for arbitrary , thereby bridging the previously reported trends in thin and overlapping double layer limits [27, 28, 2, 36]. Now, substituting Eqs. (18) and (19) into Eq. (21), we obtain
| (22) |
The boundary conditions for Eq. (22) include and , where , i.e., the centerline potential to the right of the SDL-pore interface, is to be determined.
III.3 Transition Region
We now focus on the transition region between the SDL and the mouth of the pore. First, we emphasize that the transition region is only relevant for overlapping double layer limits. In the thin double layer limit, inside the majority of the pore as well as the SDL region, and thus the transition region becomes irrelevant. This is consistent with the derivations in Biesheuvel et al. [2, 30], who assume a continuous variation in across the SDL-pore interface in the thin double layer limit, thereby implicitly neglecting the transition region.
In contrast, in the overlapping double layer limit, inside the pore is non-zero whereas in the SDL region is zero, and a transition region is required to connect the two regions. Accordingly, inside the transition region, varies from zero to the value inside the pore. To estimate the thickness of the transition region (scaled by ), we emphasize that charge density is related to the length scale of the charge gradients by the Poisson equation; see Eq. (2c). Because the radial variation inside the SDL can be ignored, the dimensionless length scale over which charge gradients could be present inside the SDL is . Therefore, using Eq. (2c) and assuming , it is straightforward to show that , enabling us to assume electroneutrality inside the SDL. In contrast, the smallest length scale over which the charge gradients are present inside the pore is , which implies [36, 49, 44, 50, 51]; see also Eq. (29). Next, in the transition region, varies from zero to the value inside the pore. Therefore, inside the transition region is on the same order of magnitude as that inside the pore. Accordingly, the relevant length scale is the same as that of the pore, or . In summary, the transition region is thin due to geometrical features of the pore.
In the limit of , the transition region may present finite electric potential and charge density changes [36]. Therefore, in our perturbation expansion model, we approximate this region as a jump discontinuity. Defining and , the charge flux in the SDL is given by where ; see Eqs. (2e) and (9). In contrast, the charge flux inside the pore is given by ; see Eqs. (2d), (18) and (19). Therefore, since the transition region is thin and thus has negligible charge storage, we ensure current conservation across it by requiring , i.e.,
| (23) |
Eq. (23) consists of two variables, i.e., and , and we need an additional equation to solve .
To relate and , we require current conservation also on the left surface of the transition region (see Fig. 1b), relating its charge flux to that of the SDL. To do so, we define and . The charge flux inside the transition region can be approximated by with a error, where is the dimensionless thickness of the region, scaled by . The current conservation relation, , reads
| (24) |
For , the right-hand side can be neglected, so . Eq. (17) can then be used to derive
| (25) |
Eq. (25) shows that when , , which is expected for thin double layers. In contrast, when , , which is also expected, since the potential will be close to the surface potential everywhere inside the pore. Substituting Eq. (25) in Eq. (23) yields
| (26) |
where is the Biot number.
III.4 Governing Equation
We can now combine Eqs. (22) and (26) to rewrite the governing equation for centerline potential as
| (27a) | |||
| where and . Here, and are the effective dimensionless time and potential, respectively. Eq. (27a) is subjected to , and . The analytical solution of Eq. (27a) yields [44] | |||
| (27b) | |||
| where . Eq. (27b) can be used to derive , which can then be used to evaluate and inside the pore using Eqs. (18) and (19). Finally, Eqs. (9) and (25) enable us to evaluate inside the SDL. | |||
We emphasize that Eqs. (18), (19) and (27c) are the key result of this paper. To the best of our knowledge, this is the first solution of the charge density and potential profiles within a cylindrical pore for arbitrary pore size in the limit of low potentials, formally justified by a regular perturbation expansion. This paper highlights that the mathematical structure of remains identical irrespective of . However, the effect of modifies the charging timescale
| (27c) |
In the thin-double-layer limit, i.e., , , consistent with the results reported previously [2]. In the overlapping-double-layer limit, i.e., , , consistent with the results reported in Gupta et. al. [36]. We illustrate this timescale dependence on pore size with contour plots of the time evolution of the electric potential in Fig. 2, and rationalize its mechanism in the following discussion.
In the thin-double-layer limit, the pore remains uncharged except in the vicinity of its surface. As shown in Fig. 2 and the Supplementary Video, for , displays a charge density profile very close to that of , indicating an earlier saturation of the profile and therefore a lower timescale. This is also backed up by the calculations, which predict for ; see Eq. (27c). In contrast, when double layers are not thin, i.e., , a radial distribution charge forms throughout the pore, producing a transient axial gradient of charge. The charge profiles continue to develop in both radial and axial directions and appear to saturate around . This is also consistent with our prediction since ; see Eq. (27c). The charging of lies between the two other scenarios described. Finally, we also note that while the overlapping double layers take longer to charge, they also store more charge throughout the pore, resulting in a trade-off of charge density and charging timescale.
The characteristic feature that sets the charging timescale of arbitrary pore sizes is the interplay of electromigrative and diffusive fluxes. Therefore, we construct a vector plot of diffusive and electromigrative fluxes by employing our analytical derivation; see Fig. 3. First, we note that for both and , the radial diffusive flux is always balanced by the radial electromigrative flux, even though the radial diffusive and electromigrative fluxes increase with time; see Fig. 3. This is a classical feature of double-layer charging; see Ref. [11] for more details. Overall, the balance in the radial direction implies that the timescale of pore charging is controlled by the fluxes in the axial direction. For , the axial gradient of charge vanishes and electromigration is the only mechanism promoting axial transport of charge, in consonance with the literature [27, 28, 2, 30]. In contrast, for , both electromigration and diffusion cooperate in driving charge transport; see Fig. 3. However, this increase in charge flux of in the overlapping double layer limit is smaller than the boost in charge density, leading to a longer charging timescale.


IV Results: Potential, Charge and Current




In this section, we analyze the analytical predictions of the electrical potential and charge density profiles. We also validate our analytical predictions with the results from DNS.
IV.1 Axial Dependence of Potential and Charge Profiles
The centerline potential, given by Eq. (27b) with , and the charge density distribution, given by Eq. (17), are shown in Fig. 4. We note that we obtain excellent agreement between the analytical results and DNS for both and .
Figs. 4b and 4d show that the centerline charge density increases monotonically as is reduced, vanishing in the thin-double-layer regime, and approaching twice the applied potential for overlapping double layers. At steady state, , , and ; see Eq. (17). The increase in centerline charge induces a larger potential change at the SDL-pore transition in order to balance diffusion and electromigration in this region.
IV.2 Radial Dependence of Potential and Charge Profiles
The radial dependence of the pore potential and charge distribution, given by Eqs. (18) and (19), are shown in Fig. 5 for . The charge distribution propagates gradually along the -direction, being larger near the mouth of the pore. The potential and charge distributions are related in the radial direction in such a way that the net radial flux vanishes for all times. Figs. 5a and 5b show opposite dependences of charge density and electric potential on the axial coordinate – decreases and increases with . Essentially, the effect of the charge flux in the SDL is to transport charge into the pore, which subsequently diffuses from the mouth to the end of pore. On the other hand, potential is screened, i.e., reduced, by the charge transported from the SDL. Thus, it is lower closer to the mouth of the pore and increases with . At steady state, the charge and potential distributions become independent of . Using Eqs. (18) and (19), the distributions for potential and charge throughout the pore at steady state are found to be
| (28) |
and
| (29) |




We note that smaller implies higher charge density throughout the pore. However, we reiterate that this comes at the cost of a higher charging timescale, since .
IV.3 Dependence of Charge Flux on Relative Pore Size
The centerline potential gives an important physical descriptor of the charging process in the dimensionless charge flux.
Eqs. (2e), (25), and (27b) can be utilized to show that
| (30) |
The charge flux comes out to be initially independent of the relative pore size since double layers have not yet formed in the initial state, and the SDL is subjected to the potential gradient imposed by the applied potential; see Eq. (3). The effect of is thus to control the charging timescale. In fact, recall that and note that , alternatively demonstrating that narrower pores take longer to charge. This is illustrated in Fig. 6, which shows exponential-like behavior at large times, with steeper descents for larger .

IV.4 Dependence of Charge Flux on the Biot Number
The ratio of diffusion coefficients in the static diffusion layer and in the pore, , may not be unity. This might be due to different diffusion mechanisms in the SDL and the pore, i.e., bulk diffusion in the former vs. Knudsen diffusion in the latter due to its narrow size [52]. The influence of this ratio is encompassed in the Biot number, , which in this case is a ratio of charge transport resistance in the pore vs. in the SDL. Fig. 7 illustrates the dependence of pore charging on the Biot number. It shows that the higher the Biot number, the lower the resistance to charge transfer in the SDL, producing an intuitive effect: enhancement of charge flux for short-times (e.g., owing to higher SDL diffusivity), but also quicker saturation of the pore charge storage. Thus, the order of the curves for different Biot numbers swap over time. We also find that the long-time charging is given by the dominant timescale , where is the first eigenvalue satisfying the characteristic equation . This is illustrated by the good agreement between the charge fluxes and their approximation by the first mode in the Fourier series of Eq. (30), represented by dash-dotted lines. It should be noted that net charge storage is not influenced by , as will be shown in Sec. 5.2.

IV.5 Validity for Higher Potentials
For higher applied potentials, the full PNP equations assuming radial equilibrium and relaxing the constraint of constant salt follow from Eqs. (2) as
| (31) |
| (32) |
The main simplification that comes from the linear regime is the neglect of the term , whence the salt transport equation resumes to a transient diffusion equation. Given the initial and boundary conditions for salt, the trivial solution holds, i.e., we can assume salt to be constant. For higher potentials, though, that approximation ceases to be valid and the variation of salt density in the domain influences the strength of electromigration of charge.
In order to assess the validity of our analysis, beyond which the neglect of higher-order corrections in becomes unwarranted, we compare our results to the DNS for different applied potentials in Fig. 8. We obtain good agreement between potentials and charge profiles until the applied potential reaches approximately twice the thermal potential, or approximately 50 mV at room temperature. Above that threshold, a non-linear potential response in the SDL is observed in the DNS, and thus the first-order corrections underpredict the potential in the pore and overpredict the charge stored. Indeed, Fig. 8e shows that the charge flux predictions agree with DNS results for early times (), even for moderately high applied voltages, with an error of 8% for 200 mV (but of 15% for 100 mV). However, for late times, non-linear terms become important, introducing electromigration of salt which can affect the transport of charge.



V Analysis of Capacitance
Several studies on electrode charging invoke transmission line representations, theoretically developed for thin double layers [27, 28, 29], to address electric potential and capacitance in these systems [22, 23, 24, 25]. In this section, we demonstrate that a similar transmission line model can be constructed for arbitrary pore sizes. However, a time-dependent transition-region resistor makes it challenging to compare it directly with experiments. We also derive an effective capacitance for arbitrary pore sizes from the steady-state charge density profiles.
V.1 Transmission Line Circuit
First, we derive the value of conductivity for arbitrary . Briefly restoring dimensions and utilizing Eqs. (2e), (18) and (19), we find that axial charge flux inside the pore is given as
| (33) |
such that dimensional pore conductivity is given by . Note that the conductivity for is only based on electromigrative charge flux. In contrast, the conductivity for is enhanced because both diffusive and electromigrative fluxes contribute to current; see Fig. (3).
Next, we derive the dimensional capacitance per unit surface area, . We note that should yield the total charge stored per unit axial length. By utilizing Eq. (19) and integrating along the radial direction, it is straightforward to show that . In the thin-double-layer limit , we recover the usual expression that . In contrast, in the thick-double-layer limit , we obtain . This result is intuitive since it demonstrates that the length scale of capacitance in the overlapping-double-layer limit is controlled by the pore size. Based on these expressions, we can also recover , consistent with Eq. (27c).
The linear relation between average diffusive and electromigrative axial fluxes allowed us to determine expressions for pore capacitance and resistance that satisfy Ohm’s law and the definition of a capacitive element. However, the key distinguishing feature from the classical thin double layer analysis [27, 28] is the inclusion of potential change across the transition region as derived in Eq. (25), which comes from the charge flux matching. In order to account for this change in potential, we add a resistor representing the interface, as shown in Fig. V.1a. Its dimensional resistance is determined from Ohm’s law,
| (34) |
We derive an expression for dimensionless transition-region resistance in Appendix A along with the traditional expression for SDL resistance, since their final expressions are not utilized for further analysis.
Figs. V.1b and V.1c show the pore capacitance and charging time as per the transmission line model. The sharp increase in pore capacitance per unit surface area for smaller may suggest a blowup of charge flux for overlapping double layers, which is not observed in Fig. 6. In fact, though pore capacitance increases, pore resistance decreases commensurately, in a way that yields for overlapping double layers. Even more importantly, pore capacitance of arbitrary reported in our manuscript is useful for the transmission circuit analysis that predicts centerline potential, but since it’s based on the potential difference , it is not representative of experimental measures of capacitance, which are based on [3, 43]. This is the motivation for the definition of an effective capacitance that we develop in the next section.
#



V.2 Macroscopic Perspective
Despite the possibility of representing pore charging by the transmission line circuit described in Sec. V.1, its time-dependent transition-region resistor enters as an additional factor influencing the pore charging performance. In addition, the definition of pore capacitance based on the potential difference makes it incompatible with experimental measures. Therefore, in the interest of facilitating the analysis of pore-size effects on charging performance, our focus lies on a direct comparison via a macroscopic perspective, i.e., from characterizing the total charge stored by the pore at steady state. We employ the definition of effective volumetric capacitance as the ratio of total charge stored per applied voltage and unit volume. Going back to dimensionless variables and performing a radial integration of the steady-state charge density profile of Eq. (29), it is straightforward to obtain
| (35) |
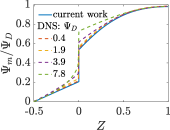
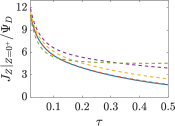
where capacitance is scaled by . Eq. (35) shows that , i.e., dimensionless volumetric capacitance is equal to dimensionless charging time; see Figs. V.1c and 10. The latter shows that the volumetric capacitance decreases with an increase in relative pore size, that is, overlapping double layers present optimal energy storage. A similar behavior of energy density increase with width reduction due to a decrease of the electroneutral region has been reported in Refs. [53] for nanopores, and [33] for pores ranging from 0.5 to 10 nm. However, in our model, this comes at a cost of a corresponding increase in the charging time . From an engineering perspective, this prediction means that an increase in the energy density, , with a change in relative pore size is unaccompanied by changes in power density, ; see the inset in Fig. 10. In fact, this relationship between energy and power density is consistent with experiments; see Fig. 1 of Ref. [6]. To the best of our knowledge, this interplay between energy and power density and its dependence on pore sizes hasn’t been derived previously.

V.3 Effect of Pore-Size Distribution
An important advantage of our analysis is that it is valid for an arbitrary , thus it can also be conducted for distributions of pore sizes to study the impact of polydispersity. Within the limitations of our model, i.e., specificity to non-interacting pores, we propose a simplified model to examine the influence of double-layer thickness over electrode charging. To this end, we assume a log-normal probability distribution function of pore sizes, in consonance with experimentally measured pore-size distributions [54, 55], and perform averages of the pore properties described in Sec. V.2 over the pore-size distribution; see Appendix C. In our analysis, we determine the effects of pore-size average and polydispersity. Fig. 11 shows that distributions with lower averages or polydispersities of relative pore sizes present higher average capacitances (i.e., the electrode capacitance) due to elevated volumetric capacitance of pores with low relative pore size. Though this conclusion is specific to the probability density function employed, the principle of boosting average capacitance with regards to double layer thickness by increasing the frequency of narrow pores should hold in general. Optimal energy density for monodisperse pore distributions has also been reported in Monte Carlo simulations of nanopores [53]. Nevertheless, the one caveat that also follows from our analysis is the accompanying increase in electrode charging time.


There is an ongoing debate about the experimentally observed behavior of capacitance for sub-nanometer pores. While some studies report an anomalous areal capacitance (i.e., capacitance per unit area) increase in sub-nanometer pores [56, 57, 6], other works claim that it is roughly independent of pore size in that regime[58, 59], the discrepancy being attributed to inaccuracies in BET isotherm surface area determination for subnanometer pores. The results of our work seem to support the latter hypothesis, showing only a mild increase of areal capacitance with pore size for (corresponding to nm for 1 M electrolytes at room temperature); see Appendix A for calculations details. However, we acknowledge that our model may fail to capture intricacies of subnanometer pores, such as anomalous capacitance increases due to loss of solvation shells [57, 6]. This is expected since the Poisson-Nernst-Planck equations do not take into account finite ion-radius and confinement effects [60], which will become crucial in the subnanometer regime.
VI Conclusion
In summary, in this article:
-
•
A regular perturbation expansion model for double layer charging at arbitrary pore sizes is proposed. The effects of arbitrary pore size include a charge flux matching condition that sets the potential change at the pore-SDL transition region;
- •
-
•
Physical insight into the mechanisms setting capacitance and charging timescale of pores with arbitrary sizes is obtained: the influence of electromigration and charge diffusion is quantified;
-
•
Electrical-double-layer charging for arbitrary pore sizes can be represented in the form of a transmission line circuit, but with the inclusion of a time-dependent interfacial capacitance. To mitigate this complexity, Eq. (30) should be utilized to calculate charge flux;
- •
Our methodology provides valuable insight into the effects of electromigration and diffusion in double-layer charging for arbitrary pore sizes. For thin double layers, electromigrative flux controls the charge flux and the charging timescale. In contrast, for overlapping double layers, the diffusive and electromigrative fluxes cooperate, enhancing the total charge flux. Yet, this increase in flux for overlapping double layers is less than the corresponding boost in charge density. This leads to a longer charging timescale in narrow pores, and thus a trade-off between charge stored and charging timescale. We also report a simplified model example of the influence of double layer thickness over electrode charging for non-interacting pores via phenomenological averages under a proposed log-normal distribution. In this case study, the same single-pore proportional increases of capacitance and charging timescale manifest in a distribution of isolated pores, predicting a constant electrode power density regardless of relative pore size.
Our approach can be extended to studying hybrid supercapacitors by adding reactions to the boundary conditions. The perturbation expansion analysis proposed here can also be utilized for asymmetric ionic valences [19] and diffusivities [61, 41, 42, 62], scenarios which are commonly observed in electrochemical devices. Finally, to compare directly with cyclic voltammetry data obtained from experiments, a similar approach can also be employed for higher and/or time-dependent applied potentials.
Conflicts of Interest
There are no conflicts to declare.
Acknowledgements
The authors would like to acknowledge the helpful input provided by Howard A. Stone, Daniel K. Schwartz, Adam Holewinski, Gesse Roure, Nathan Jarvey, and anonymous Referee #2. P.J.Z. would like to acknowledge the support of a project that has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 847413 and was a part of an international co-financed project founded from the program of the Minister of Science and Higher Education entitled “PMW” in the years 2020–2024; agreement No. 5005/H2020-MSCA-COFUND/2019/2. The authors acknowledge that the simulations reported in this contribution were performed using the Princeton Research Computing resources at Princeton University which is consortium of groups including the Princeton Institute for Computational Science and Engineering and the Princeton University Office of Information Technology’s Research Computing department.
References
- Becker [1957] H. I. Becker, Low voltage electrolytic capacitor (U.S. Patent 2800616A, United States Patent and Trademark Office, 1957).
- Biesheuvel and Bazant [2010] P. Biesheuvel and M. Bazant, Nonlinear dynamics of capacitive charging and desalination by porous electrodes, Phys. Rev. E 81, 031502 (2010).
- Zhang et al. [2014] C. Zhang, K. B. Hatzell, M. Boota, B. Dyatkin, M. Beidaghi, D. Long, W. Qiao, E. C. Kumbur, and Y. Gogotsi, Highly porous carbon spheres for electrochemical capacitors and capacitive flowable suspension electrodes, Carbon 77, 155 (2014).
- Presser et al. [2012] V. Presser, C. R. Dennison, J. Campos, K. W. Knehr, E. C. Kumbur, and Y. Gogotsi, The electrochemical flow capacitor: A new concept for rapid energy storage and recovery, Adv. Energy Mater. 2, 895 (2012).
- Winter and Brodd [2004] M. Winter and R. J. Brodd, What are batteries, fuel cells, and supercapacitors?, Chem. Rev. (Washington, DC, U. S.) 104, 4245 (2004).
- Simon and Gogotsi [2008] P. Simon and Y. Gogotsi, Materials for electrochemical capacitors, Nat. Mater. , 845 (2008).
- Muzaffar et al. [2019] A. Muzaffar, M. B. Ahamed, K. Deshmukh, and J. Thirumalai, A review on recent advances in hybrid supercapacitors: Design, fabrication and applications, Renewable Sustainable Energy Rev. 101, 123 (2019).
- Gouy [1910] M. Gouy, Sur la constitution de la charge électrique à la surface d’un électrolyte, J. Phys. Theor. Appl. 9, 457 (1910).
- Chapman [1913] D. L. Chapman, Li. a contribution to the theory of electrocapillarity, The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science 25, 475 (1913).
- Stern [1924] O. Stern, Zur theorie der elektrolytischen doppelschicht, Zeitschrift Für Elektrochemie Und Angewandte Physikalische Chemie 30, 508 (1924).
- Bazant et al. [2004] M. Z. Bazant, K. Thornton, and A. Ajdari, Diffuse-charge dynamics in electrochemical systems, Phys. Rev. E 70, 021506 (2004).
- Feicht et al. [2016] S. E. Feicht, A. E. Frankel, and A. S. Khair, Discharging dynamics in an electrolytic cell, Phys. Rev. E 94, 012601 (2016).
- Kilic et al. [2007a] M. S. Kilic, M. Z. Bazant, and A. Ajdari, Steric effects in the dynamics of electrolytes at large applied voltages. i. double-layer charging, Phys. Rev. E 75, 021502 (2007a).
- Kilic et al. [2007b] M. S. Kilic, M. Z. Bazant, and A. Ajdari, Steric effects in the dynamics of electrolytes at large applied voltages. ii. modified poisson-nernst-planck equations, Phys. Rev. E 75, 021503 (2007b).
- Balu and Khair [2018] B. Balu and A. S. Khair, Role of stefan–maxwell fluxes in the dynamics of concentrated electrolytes, Soft Matter 14, 8267 (2018).
- Jiménez-Ángeles et al. [2006] F. Jiménez-Ángeles, G. Odriozola, and M. Lozada-Cassou, Electrolyte distribution around two like-charged rods: Their effective attractive interaction and angular dependent charge reversal, J. Chem. Phys. 124, 134902 (2006).
- Jiménez-Ángeles and Lozada-Cassou [2008] F. Jiménez-Ángeles and M. Lozada-Cassou, On the regimes of charge reversal, J. Chem. Phys. 128, 174701 (2008).
- Wu et al. [2011] J. Wu, T. Jiang, D.-e. Jiang, Z. Jin, and D. Henderson, A classical density functional theory for interfacial layering of ionic liquids, Soft Matter 7, 11222 (2011).
- Gupta and Stone [2018] A. Gupta and H. A. Stone, Electrical double layers: effects of asymmetry in electrolyte valence on steric effects, dielectric decrement, and ion–ion correlations, Langmuir 34, 11971 (2018).
- Gupta et al. [2020a] A. Gupta, A. G. Rajan, E. A. Carter, and H. A. Stone, Ionic layering and overcharging in electrical double layers in a poisson-boltzmann model, Phys. Rev. Lett. 125, 188004 (2020a).
- Gupta et al. [2020b] A. Gupta, A. Govind Rajan, E. A. Carter, and H. A. Stone, Thermodynamics of electrical double layers with electrostatic correlations, J. Phys. Chem. C 124, 26830 (2020b).
- Black and Andreas [2010] J. M. Black and H. A. Andreas, Pore shape affects spontaneous charge redistribution in small pores, J. Phys. Chem. C 114, 12030 (2010).
- Kaus et al. [2010] M. Kaus, J. Kowal, and D. U. Sauer, Modelling the effects of charge redistribution during self-discharge of supercapacitors, Electrochim. Acta 55, 7516 (2010).
- Kowal et al. [2011] J. Kowal, E. Avaroglu, F. Chamekh, A. Šenfelds, T. Thien, D. Wijaya, and D. U. Sauer, Detailed analysis of the self-discharge of supercapacitors, J. Power Sources 196, 573 (2011).
- Madabattula and Kumar [2018] G. Madabattula and S. Kumar, Insights into charge-redistribution in double layer capacitors, J. Electrochem. Soc. 165, A636 (2018).
- Lian et al. [2020] C. Lian, M. Janssen, H. Liu, and R. van Roij, Blessing and curse: how a supercapacitor’s large capacitance causes its slow charging, Phys. Rev. Lett. 124, 076001 (2020).
- De Levie [1963] R. De Levie, On porous electrodes in electrolyte solutions: I. capacitance effects, Electrochim. Acta 8, 751 (1963).
- De Levie [1964] R. De Levie, On porous electrodes in electrolyte solutions—iv, Electrochim. Acta 9, 1231 (1964).
- Janssen [2021] M. Janssen, Transmission line circuit and equation for an electrolyte-filled pore of finite length, Phys. Rev. Lett. 126, 136002 (2021).
- Biesheuvel et al. [2011] P. Biesheuvel, Y. Fu, and M. Z. Bazant, Diffuse charge and faradaic reactions in porous electrodes, Phys. Rev. E 83, 061507 (2011).
- Peters et al. [2016] P. Peters, R. Van Roij, M. Z. Bazant, and P. Biesheuvel, Analysis of electrolyte transport through charged nanopores, Phys. Rev. E 93, 053108 (2016).
- Levy et al. [2020] A. Levy, J. P. de Souza, and M. Z. Bazant, Breakdown of electroneutrality in nanopores, J. Colloid Interface Sci. 579, 162 (2020).
- Varghese et al. [2011] J. Varghese, H. Wang, and L. Pilon, Simulating electric double layer capacitance of mesoporous electrodes with cylindrical pores, J. Electrochem. Soc. 158, A1106 (2011).
- Alizadeh and Mani [2017] S. Alizadeh and A. Mani, Multiscale model for electrokinetic transport in networks of pores, part i: model derivation, Langmuir 33, 6205 (2017).
- Alizadeh et al. [2019] S. Alizadeh, M. Z. Bazant, and A. Mani, Impact of network heterogeneity on electrokinetic transport in porous media, J. Colloid Interface Sci. 553, 451 (2019).
- Gupta et al. [2020c] A. Gupta, P. J. Zuk, and H. A. Stone, Charging dynamics of overlapping double layers in a cylindrical nanopore, Phys. Rev. Lett. 125, 076001 (2020c).
- Wiersema et al. [1966] P. Wiersema, A. Loeb, and J. T. G. Overbeek, Calculation of the electrophoretic mobility of a spherical colloid particle, J. Colloid Interface Sci. 22, 78 (1966).
- O’Brien and White [1978] R. W. O’Brien and L. R. White, Electrophoretic mobility of a spherical colloidal particle, J. Chem. Soc., Faraday Trans. 2 74, 1607 (1978).
- Yariv et al. [2011] E. Yariv, O. Schnitzer, and I. Frankel, Streaming-potential phenomena in the thin-debye-layer limit. part 1. general theory, J. Fluid Mech. 685, 306 (2011).
- Schnitzer and Yariv [2012] O. Schnitzer and E. Yariv, Macroscale description of electrokinetic flows at large zeta potentials: nonlinear surface conduction, Phys. Rev. E 86, 021503 (2012).
- Khair and Balu [2020] A. S. Khair and B. Balu, Breaking electrolyte symmetry in induced-charge electro-osmosis, J. Fluid Mech. 905, A20 (2020).
- Amrei et al. [2020] S. H. Amrei, G. H. Miller, K. J. Bishop, and W. D. Ristenpart, A perturbation solution to the full poisson–nernst–planck equations yields an asymmetric rectified electric field, Soft Matter 16, 7052 (2020).
- Boota et al. [2015] M. Boota, K. Hatzell, M. Alhabeb, E. Kumbur, and Y. Gogotsi, Graphene-containing flowable electrodes for capacitive energy storage, Carbon 92, 142 (2015).
- Deen [1998] W. M. Deen, Analysis of Transport Phenomena (Oxford University Press New York, 1998).
- Bard and Faulkner [2000] A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications (Wiley, 2000).
- Newman and Thomas-Alyea [2012] J. Newman and K. E. Thomas-Alyea, Electrochemical Systems (John Wiley & Sons, 2012).
- Weller et al. [1998] H. G. Weller, G. Tabor, H. Jasak, and C. Fureby, A tensorial approach to computational continuum mechanics using object-oriented techniques, Comput. Phys. 12, 620 (1998).
- Jasak et al. [2007] H. Jasak, A. Jemcov, Z. Tukovic, et al., Openfoam: A c++ library for complex physics simulations, in International Workshop on Coupled Methods in Numerical Dynamics, Vol. 1000 (IUC Dubrovnik Croatia, 2007) pp. 1–20.
- Chen et al. [2020] G. Chen, A. Perazzo, and H. A. Stone, Influence of salt on the viscosity of polyelectrolyte solutions, Phys. Rev. Lett. 124, 177801 (2020).
- Lyklema [2005] J. Lyklema, Fundamentals of Interface and Colloid Science: Soft Colloids, Vol. 5 (Elsevier, 2005).
- Evans and Wennerström [1999] D. F. Evans and H. Wennerström, The Colloidal Domain: Where Physics, Chemistry, Biology, and Technology Meet (Wiley-Vch New York, 1999).
- Rawlings and Ekerdt [2002] J. B. Rawlings and J. G. Ekerdt, Chemical Reactor Analysis and Design Fundamentals (Nob Hill Pub, Llc, 2002).
- Kondrat et al. [2012] S. Kondrat, C. Perez, V. Presser, Y. Gogotsi, and A. Kornyshev, Effect of pore size and its dispersity on the energy storage in nanoporous supercapacitors, Energy Environ. Sci. 5, 6474 (2012).
- Jorne and Roayaie [1986] J. Jorne and E. Roayaie, Effect of pore-size distribution on metal ion removal in flow-through porous electrodes, J. Electrochem. Soc. 133, 1649 (1986).
- Song et al. [1999] H.-K. Song, Y.-H. Jung, K.-H. Lee, and L. H. Dao, Electrochemical impedance spectroscopy of porous electrodes: the effect of pore size distribution, Electrochim. Acta 44, 3513 (1999).
- Chmiola et al. [2006] J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon, and P.-L. Taberna, Anomalous increase in carbon capacitance at pore sizes less than 1 nanometer, Science 313, 1760 (2006).
- Huang et al. [2008] J. Huang, B. G. Sumpter, and V. Meunier, Theoretical model for nanoporous carbon supercapacitors, Angew. Chem. 120, 530 (2008).
- Centeno et al. [2011] T. A. Centeno, O. Sereda, and F. Stoeckli, Capacitance in carbon pores of 0.7 to 15 nm: a regular pattern, Phys. Chem. Chem. Phys. 13, 12403 (2011).
- García-Gómez et al. [2015] A. García-Gómez, G. Moreno-Fernández, B. Lobato, and T. A. Centeno, Constant capacitance in nanopores of carbon monoliths, Phys. Chem. Chem. Phys. 17, 15687 (2015).
- Kornyshev [2007] A. A. Kornyshev, Double-layer in ionic liquids: Paradigm change?, J. Phys. Chem. B 111, 5545 (2007).
- Kim et al. [2019] J. Kim, S. Davidson, and A. Mani, Characterization of chaotic electroconvection near flat inert electrodes under oscillatory voltages, Micromachines 10, 161 (2019).
- Balu and Khair [2021] B. Balu and A. S. Khair, A thin double layer analysis of asymmetric rectified electric fields (arefs), J. Eng. Math. 129, 1 (2021).
Appendix A Transmission Line Circuit Resistances
In order to complete the transmission line circuit description in Fig. 9a, we derive the expressions of dimensional SDL and transition-region resistances. We start with the SDL resistance, which is given by Ohm’s law as
| (36) |
Utilizing Eqs. (2e) and (9), we have
| (37) |
From Eq. (25), the potential difference across the transition region is given by
| (38) |
Using Eq. (27b), it can be written as
|
Ψ_left-Ψ_right=-ΨDI0(apλ)[1-2∑_n=1^∞sin2κn2κn+sin2κnexp(-κ_n^2T)]. |
(39) |
Therefore, the dimensional transition-region resistance follows from Eq. (30) as
| (40) |
We choose the scale for the resistances, such that their dimensionless expressions read
| (41) |
and
| (42) |
Fig. (A1) shows a plot of the transition resistance over time for different relative pore sizes. As the relative pore size decreases, the resistance imposed by the transition region to the charge flux increases. This is due to the larger potential differences across the transition region for lower relative pore sizes, caused by their increased charge density.

Appendix B Areal Capacitance
Most experiments report results on a per unit surface area basis, hence we briefly present our results for areal capacitance here in order to compare them qualitatively to experiments. Denoting dimensionless areal capacitance by and scaling it by , it follows from Eq. (35) that
| (43) |
We plot this result in Fig. (A2).

Appendix C Electrode Average Properties
We note that the methodology presented here assumes non-interacting pores and overlooks pore-network configuration, pore intersections, connectivity, among others. To extend single-pore analysis for a distribution of non-interacting pores, we first consider a continuous log-normal distribution of pore sizes,
| (44) |
where the parameters and are given in terms of relative pore size average and variance by and .
Next, we formally derive the averaging procedure employed in Fig. 11. The average volumetric capacitance is defined as the total charge stored inside the electrode divided by its total volume and applied potential , compatible with what experiments denote as volumetric capacitance [43]
| (45) |
Charge is the integral of the charge density over the entire electrode volume,
| (46) |
where and are volumes of electrode material and pores, respectively. Furthermore, inside the electrode material (an ideal conductor). Therefore, the results integral can be written as a sum over all pores
| (47) |
where is the charge of a pore with volume , both known from the single-pore model. represents the total number of pores. We insert Eq. (47) into Eq. (45) and take averages, denoted by , over an ensemble of sample electrode pore configurations drawn from the same pore-size distribution. In this way, we have
| (48) |
For a non-interacting pore model, the average charge of the i-th pore only depends on its relative pore size, i.e.,
| (49) |
Substituting Eq. (49) into Eq. (48), we have
| (50) |
Next, we multiply and divide the result by the average pore volume of the electrode
| (51) |
which yields
| (52) |
Lastly, we write the charge of a pore in terms of its volumetric effective capacitance, , change variables in the integrals from to and define the electrode porosity to get
| (53) |
Next, we propose a definition of timescale for a distribution of pore sizes which reduces to a single-pore solution for a monodisperse distribution. To this end, note that Eq. (20) can be integrated over the volume of a pore to give, using Eq. (2d) and Eqs. (5) for a blocking electrode, to give
| (54) |
where and the index denotes the i-th pore. Integrating over time and performing the change of variables , we have
| (55) |
Using (54) and (55), we find that
| (56) |
Our motivation in generalizing this is to construct a measure of the total charging time of the electrode, not of the individual pores. Therefore, noting that neither the initial value of the flux nor its integral on time depend on pore size – see Eq. (30) – we opt to define the electrode charging timescale as
| (57) |
averaged over the ensemble of pore configurations, which explicitly takes the form
| (58) |
Using Eqs. (54) and (55) where and changing variables from to , we have
| (59) |
Through this approach we define a physically relevant average of charging timescale that is consistent with the single-pore definition.